关键信息
-
基因名
DNAM-1
-
简介
DNAM-1 protein is a key player in cell-cell adhesion and lymphocyte signaling, mediating cytotoxicity and lymphokine secretion of CTL and NK cells. As a receptor for NECTIN2, DNAM-1 stimulates T cell proliferation and cytokine production (IL2, IL5, IL10, IL13, and IFNG) upon ligand binding. DNAM-1 Protein, Human (HEK293, His) is the recombinant human-derived DNAM-1 protein, expressed by HEK293 , with C-6*His labeled tag.
- 应用
-
别名
DNAX accessory molecule 1; DNAM-1; CD226 antigen; CD226
-
种属
Human
-
表达系统
HEK293
-
标签
C-6*His
-
纯度
Greater than 90% as determined by SDS-PAGE.
-
蛋白编号
Q15762
-
表达区间
E19-N247
-
氨基酸序列
EEVLWHTSVPFAENMSLECVYPSMGILTQVEWFKIGTQQDSIAIFSPTHGMVIRKPYAERVYFLNSTMASNNMTLFFRNASEDDVGYYSCSLYTYPQGTWQKVIQVVQSDSFEAAVPSNSHIVSEPGKNVTLTCQPQMTWPVQAVRWEKIQPRQIDLLTYCNLVHGRNFTSKFPRQIVSNCSHGRWSVIVIPDVTVSDSGLYRCYLQASAGENETFVMRLTVAEGKTDN
-
蛋白长度
Partial
-
分子量
43-55 kDa
-
内毒素
< 1.0 EU per μg protein as determined by the LAL method.
-
性状
Freeze-dried powder
-
缓冲液
PBS, pH7.4, containing 0.01% SKL, 1mM DTT, 5% Trehalose and Proclin300.
-
复溶方法
Reconstitute in ddH2O to a concentration of 0.1-0.5 mg/mL. Do not vortex.
- 个性化定制
-
稳定性测试
The thermal stability is described by the loss rate. The loss rate was determined by accelerated thermal degradation test, that is, incubate the protein at 37℃ for 48h, and no obvious degradation and precipitation were observed. The loss rate isless than 8% within the expiration date under appropriate storage condition.
-
保存条件 & 期限
Samples are stable for up to twelve months from date of receipt at -20℃ to -80℃. Store it under sterile conditions at -20℃ to -80℃. It is recommended that the protein be aliquoted for optimal storage. Avoid repeated freeze-thaw cycles.
-
运输条件
In general, recombinant proteins are supplied as lyophilized powder and shipped at ambient temperature. For bulk packages, the proteins are provided as frozen liquid and shipped with blue ice, unless otherwise requested by the customer.
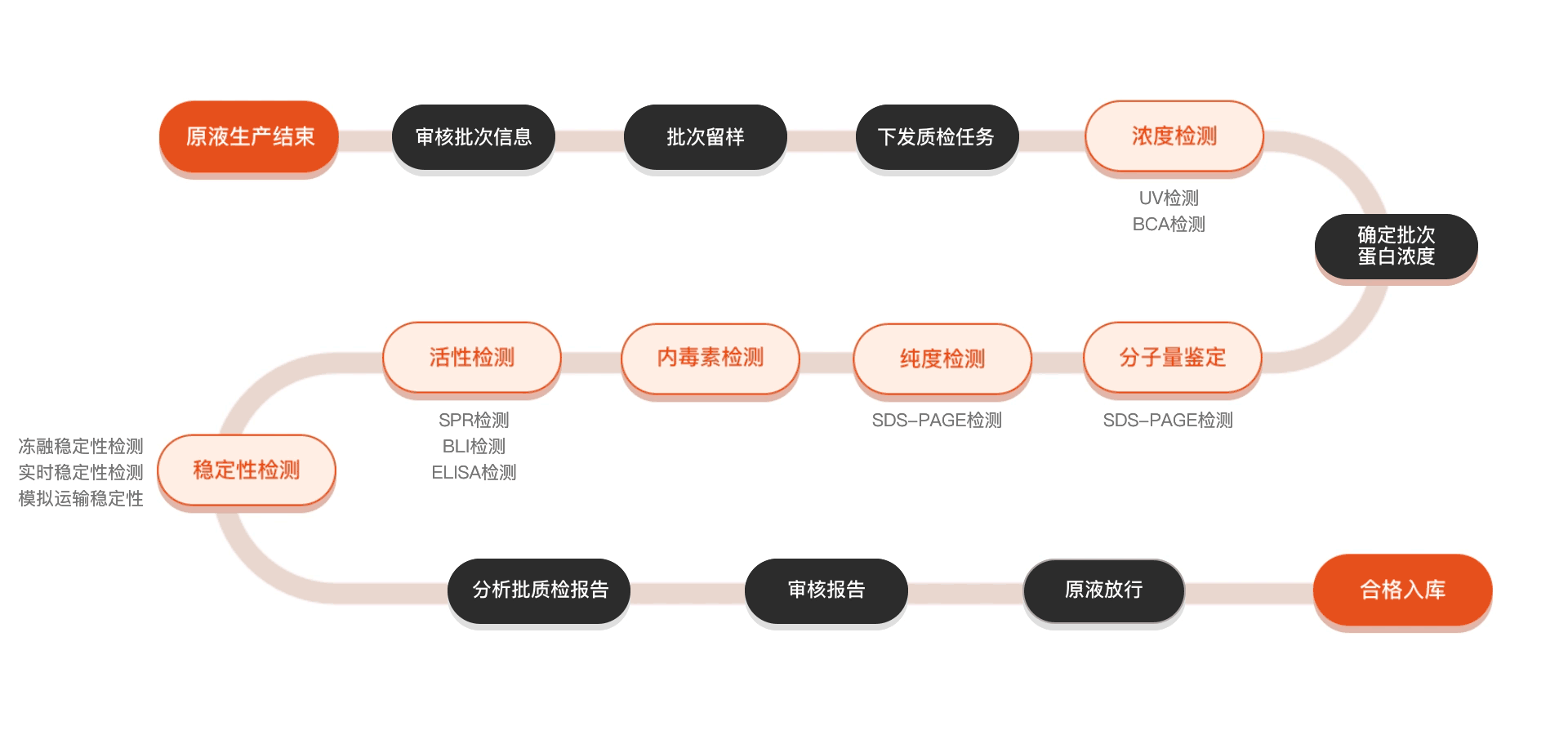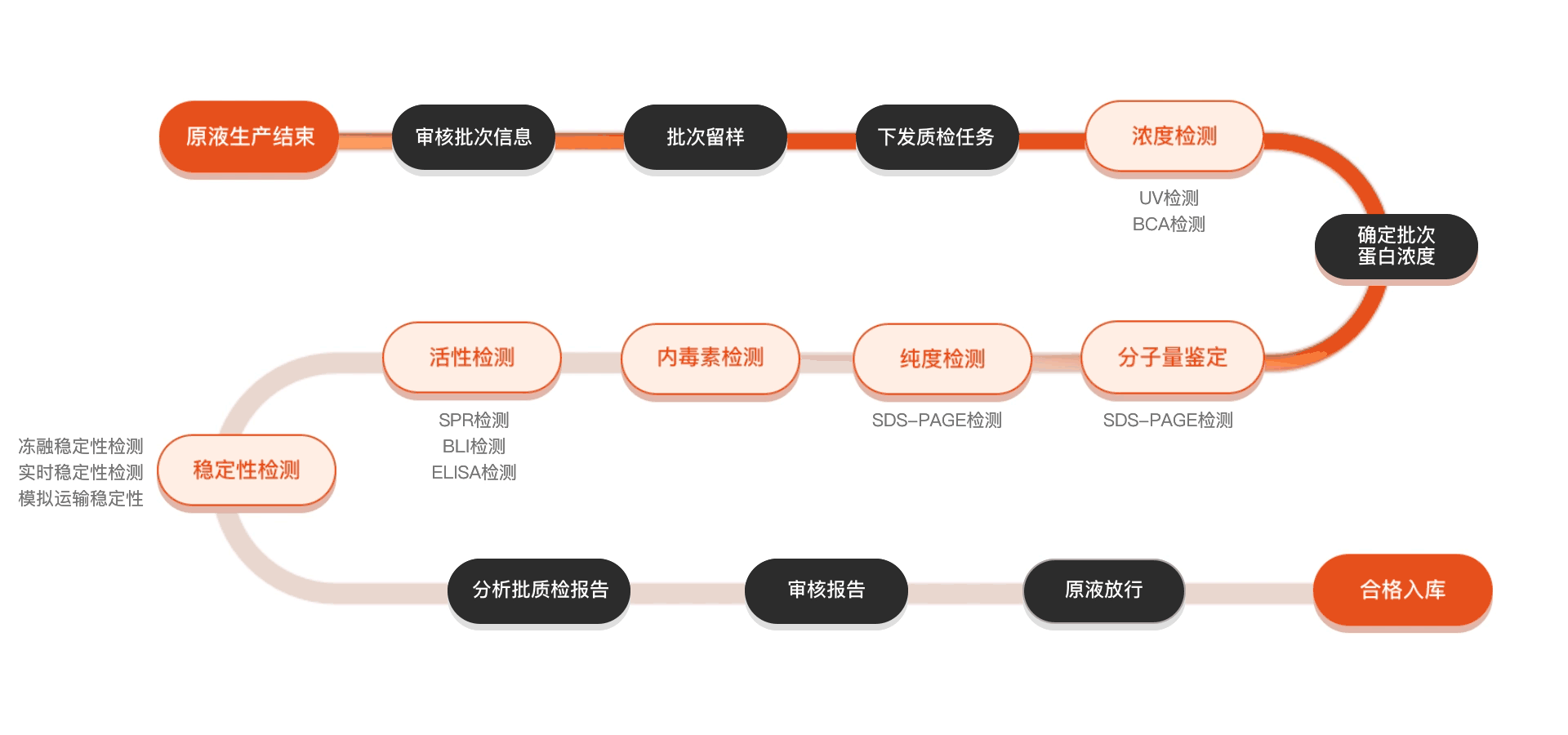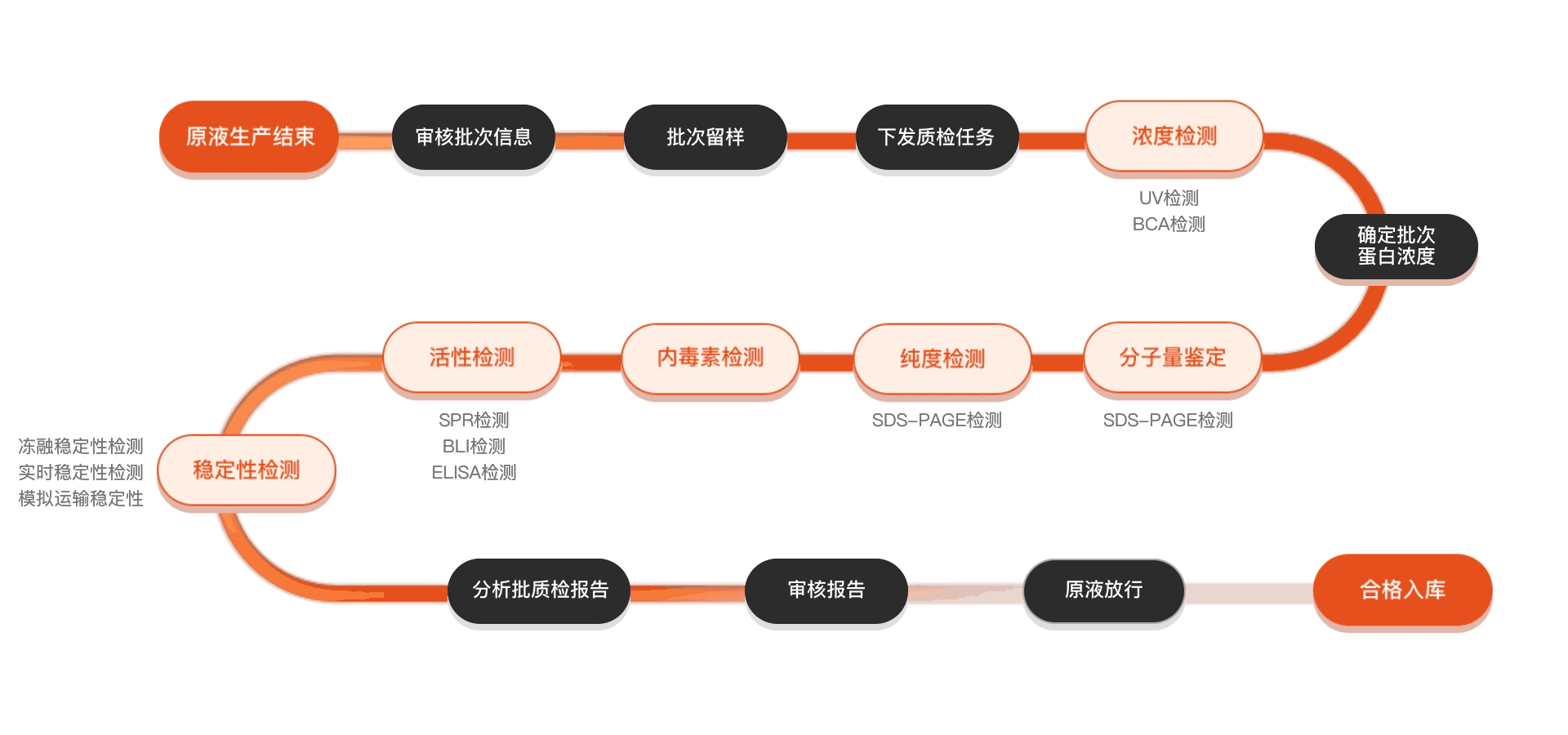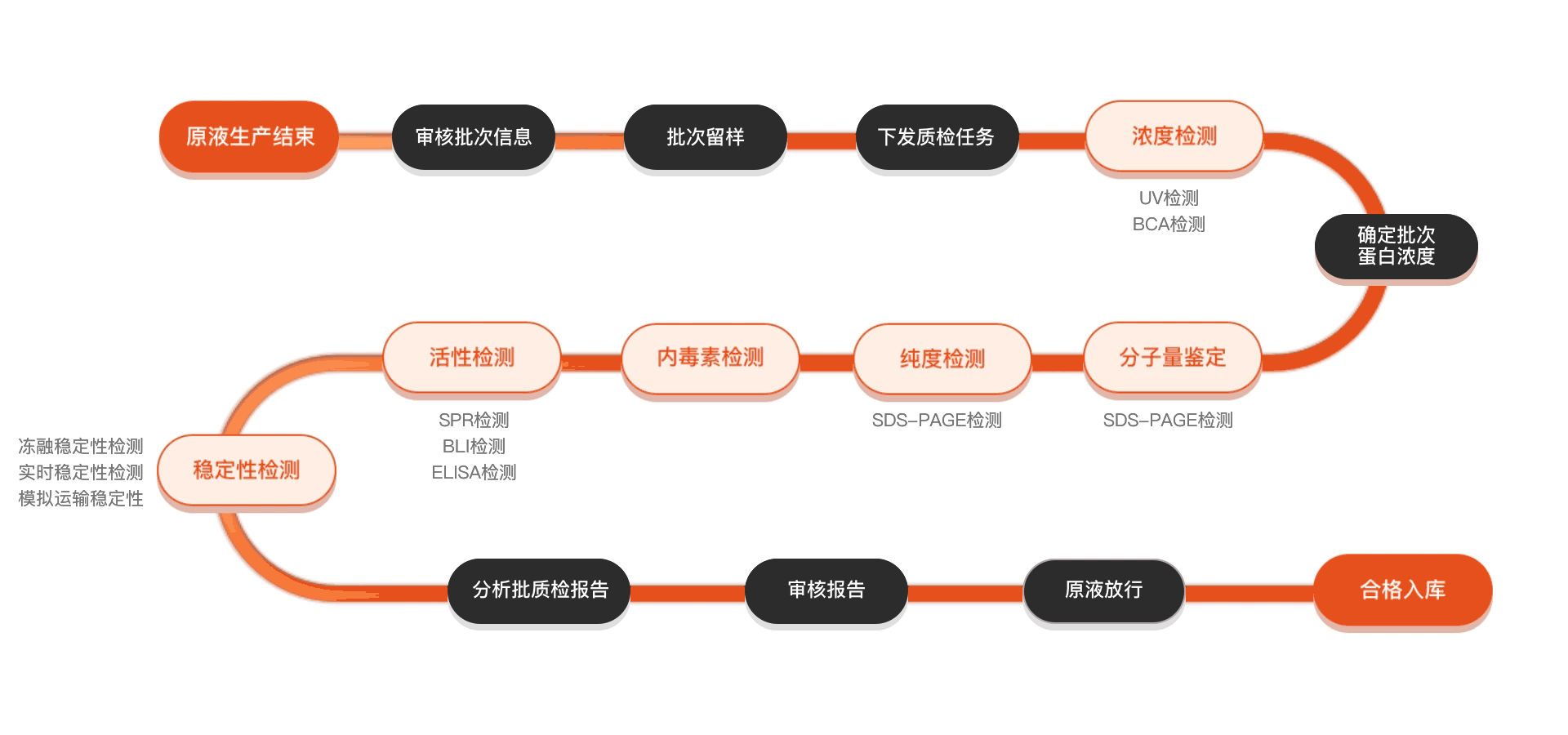
质检流程
相关产品






背景信息
CD98 is a transmembrane protein that functions as an integrin-associated protein and is involved in multiple cellular processes, including cell adhesion, proliferation, and amino acid transport. Originally identified for its role in lymphocyte activation, CD98 has garnered significant interest in cancer research due to its overexpression in various malignant tumors, suggesting its potential as a biomarker and therapeutic target. The protein consists of several key domains that facilitate its interaction with integrins and other signaling molecules, thereby influencing cell signaling pathways that drive tumorigenesis. Recent studies have focused on the structural and functional characterization of CD98 recombinant protein to elucidate its role in cancer progression and metastasis. By generating and purifying CD98 recombinant protein, researchers aim to investigate its interactions with ligands, understand its physiological functions, and explore its potential as a target for novel anti-cancer therapies. Insights gained from these studies could lead to the development of strategies that inhibit CD98 function, ultimately aiming to halt tumor growth and improve patient outcomes. Overall, the exploration of CD98 recombinant protein serves as a critical step in advancing our understanding of its biology and therapeutic potential in oncology.