关键信息
-
基因名
Nectin-1
-
简介
Nectin-1 Protein, with carbohydrate and virion binding activities, plays pivotal roles in axon guidance, synapse assembly, and viral entry. Located in dendrites, growth cone membranes, and synapses, Nectin-1 is implicated in human conditions like cleft lip and ectodermal dysplasia. Its biased expression, especially in Kidney (RPKM 45.4) and Brain (RPKM 31.3), underscores its significance in diverse physiological contexts. Nectin-1 Protein, Rat (HEK293, His) is the recombinant rat-derived Nectin-1 protein, expressed by HEK293 , with C-His labeled tag.
- 应用
-
生物活性
Measured by its binding ability in a functional ELISA. When Recombinant Human Nectin-3 is present at 1 μg/mL, can bind Recombinant Rat Nectin-1. The ED50 for this effect is 125.7 ng/mL. Measured by its binding ability in a functional ELISA. When Recombinant Human Nectin-3 is present at 1 μg/mL, can bind Recombinant Rat Nectin-1.The ED50 for this effect is 125.7 ng/mL.
-
别名
Poliovirus Receptor-Related Protein 1; Herpes Virus Entry Mediator C; Herpesvirus Entry Mediator C; HveC; Herpesvirus Ig-Like Receptor; HIgR; Nectin-1; CD111; PVRL1; HVEC; PRR1
-
种属
Rat
-
表达系统
HEK293
-
标签
C-His
-
纯度
Greater than 90% as determined by SDS-PAGE.
-
蛋白编号
F1LNP8
-
表达区间
Q31-A354
-
氨基酸序列
QVVQVNDSMYGFIGTDVILHCSFANPLPTVKITQVTWQKASNGSKQNMAIYNPTMGVSVLPPYEKRVEFLRPSFIDGTIRLSHLELEDEGMYICEFATFPTGNRESQLNLTVMAKPTNWIEGTQAVLRARKGQDDKVLVATCTSANGKPPSVVSWETRLKGEAEYQEIRNPNGTVTVISRYRLVPSREAHRQSLACIVNYHLDRFRESLTLNVQYEPEVTIEGFDGNWYLQRTDVKLTCKADANPPATEYHWTTLNGSLPKGVEAQNRTLFFRGPINYSLAGTYICEATNPIGTRSGQVEVNITEFPYTPTPEHGRRAGQMPTA
-
蛋白长度
Partial
-
分子量
50-70 kDa
-
内毒素
< 1.0 EU per μg protein as determined by the LAL method.
-
性状
Freeze-dried powder
-
缓冲液
PBS, pH7.4, containing 0.01% SKL, 1mM DTT, 5% Trehalose and Proclin300.
-
复溶方法
Reconstitute in ddH2O to a concentration of 0.1-0.5 mg/mL. Do not vortex.
- 个性化定制
-
稳定性测试
The thermal stability is described by the loss rate. The loss rate was determined by accelerated thermal degradation test, that is, incubate the protein at 37℃ for 48h, and no obvious degradation and precipitation were observed. The loss rate isless than 8% within the expiration date under appropriate storage condition.
-
保存条件 & 期限
Samples are stable for up to twelve months from date of receipt at -20℃ to -80℃. Store it under sterile conditions at -20℃ to -80℃. It is recommended that the protein be aliquoted for optimal storage. Avoid repeated freeze-thaw cycles.
-
运输条件
In general, recombinant proteins are supplied as lyophilized powder and shipped at ambient temperature. For bulk packages, the proteins are provided as frozen liquid and shipped with blue ice, unless otherwise requested by the customer.
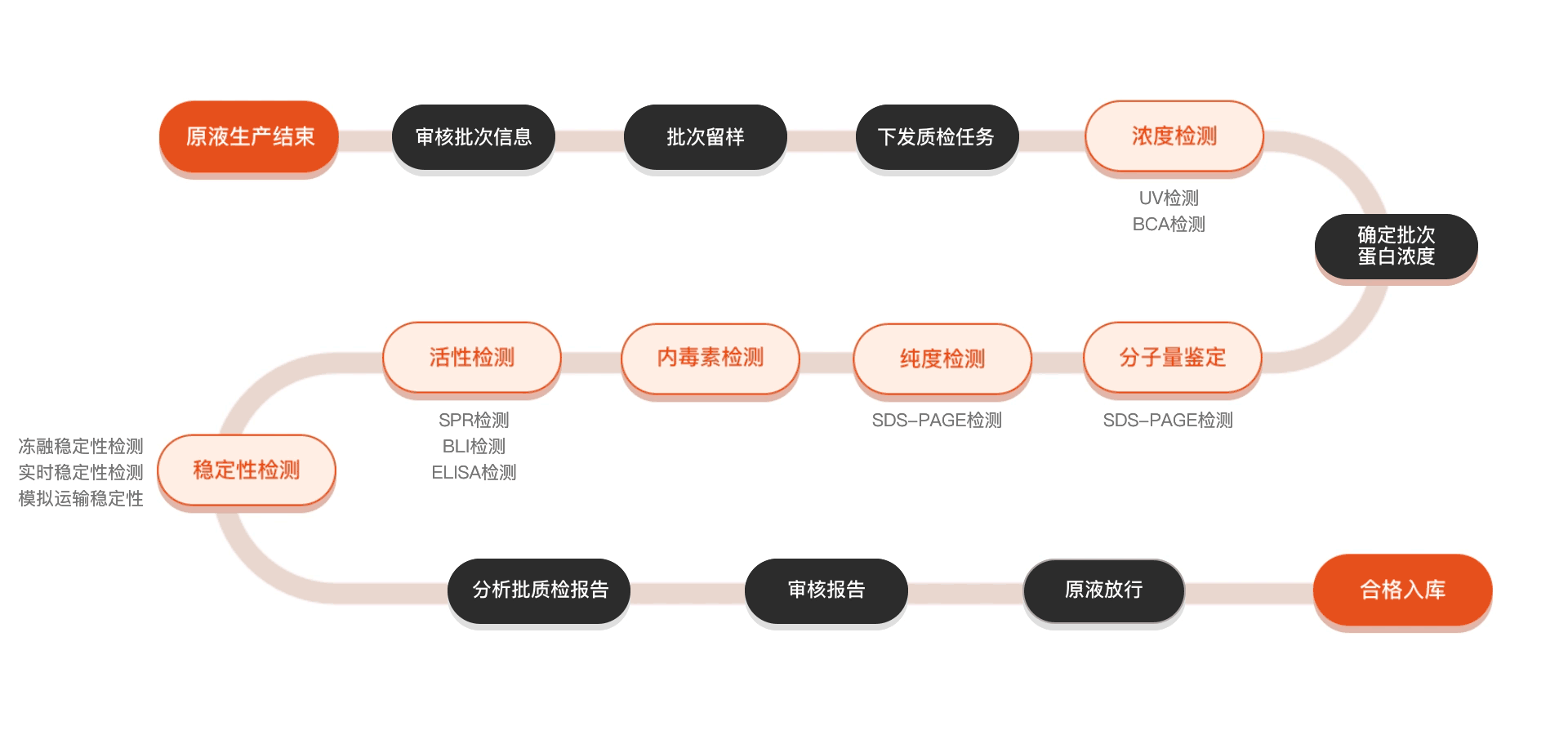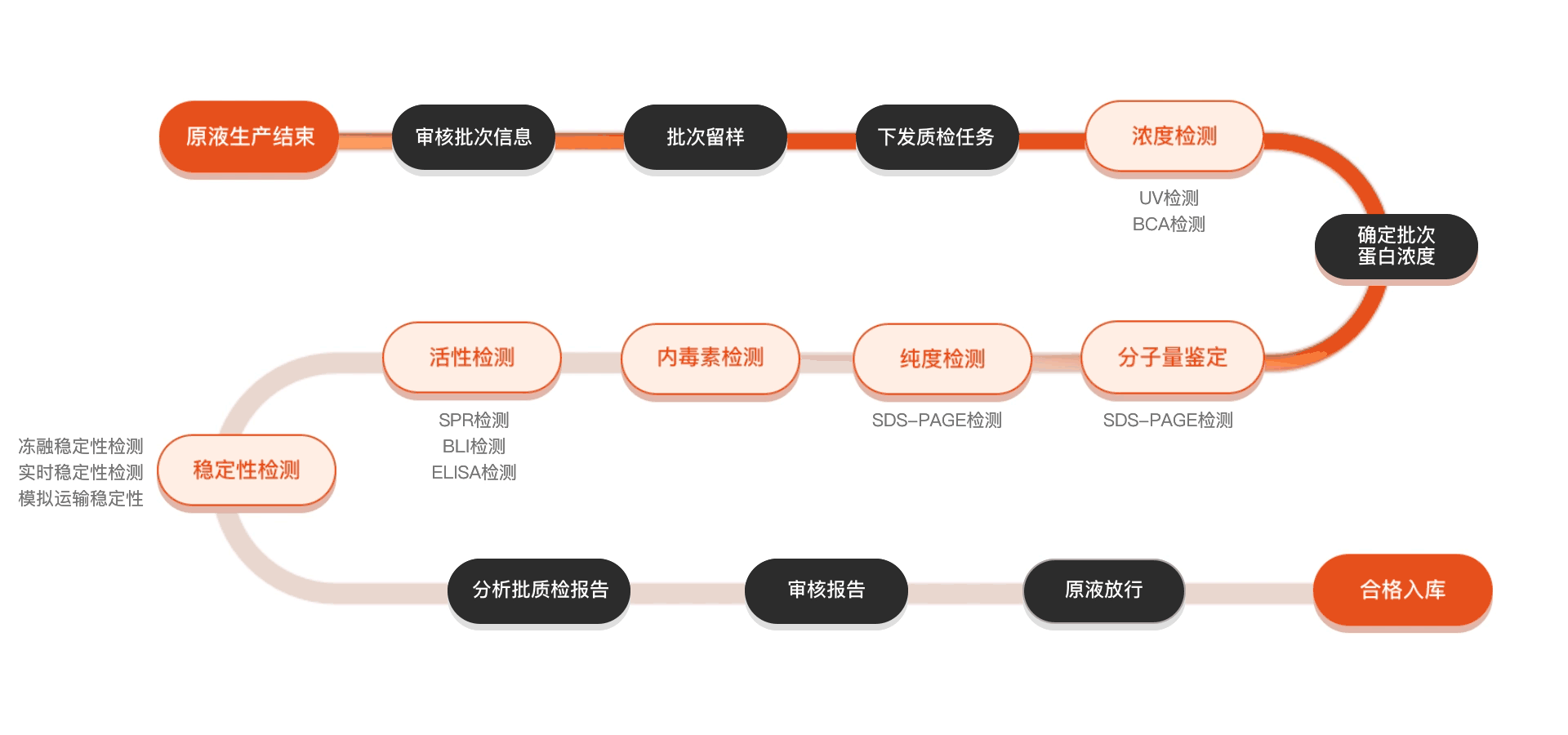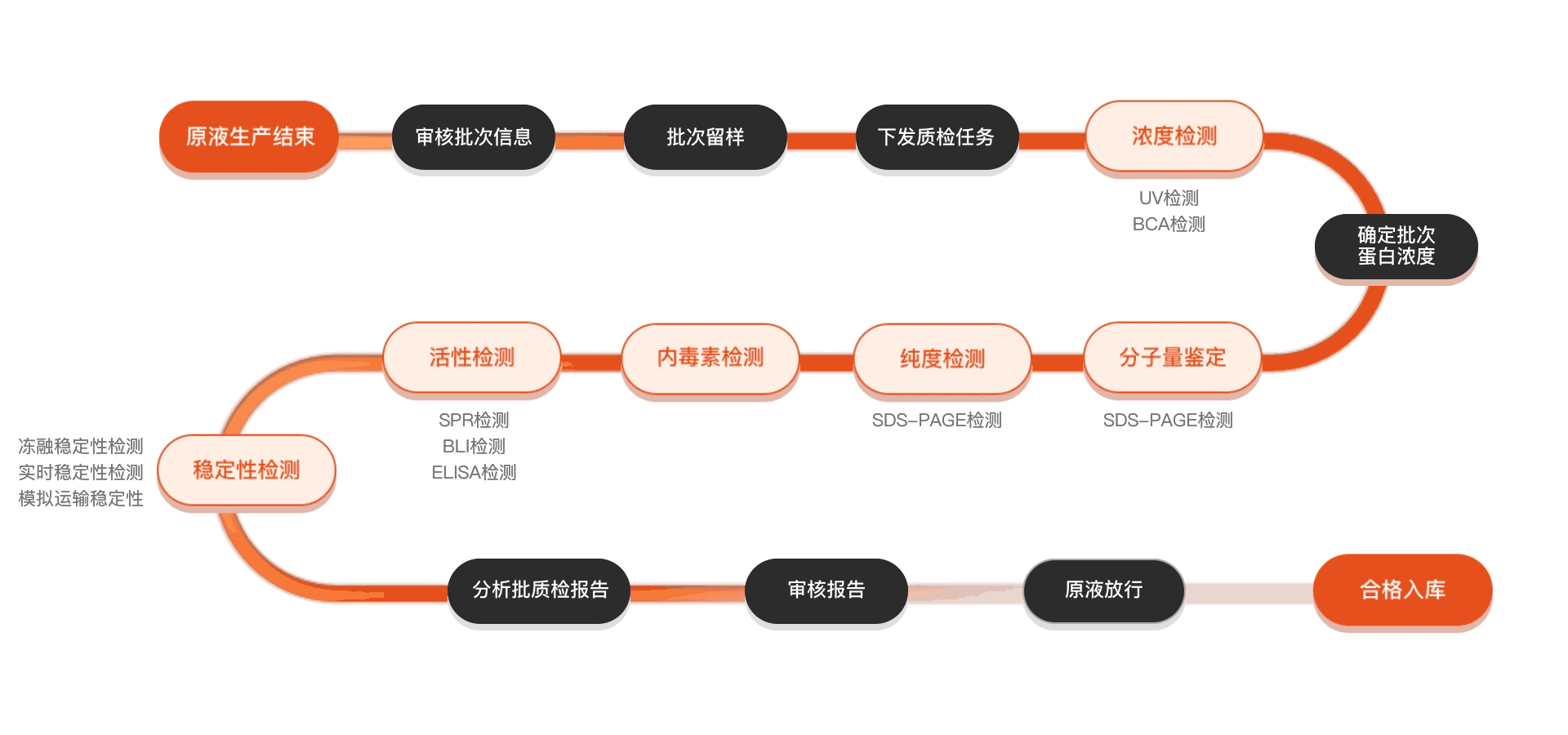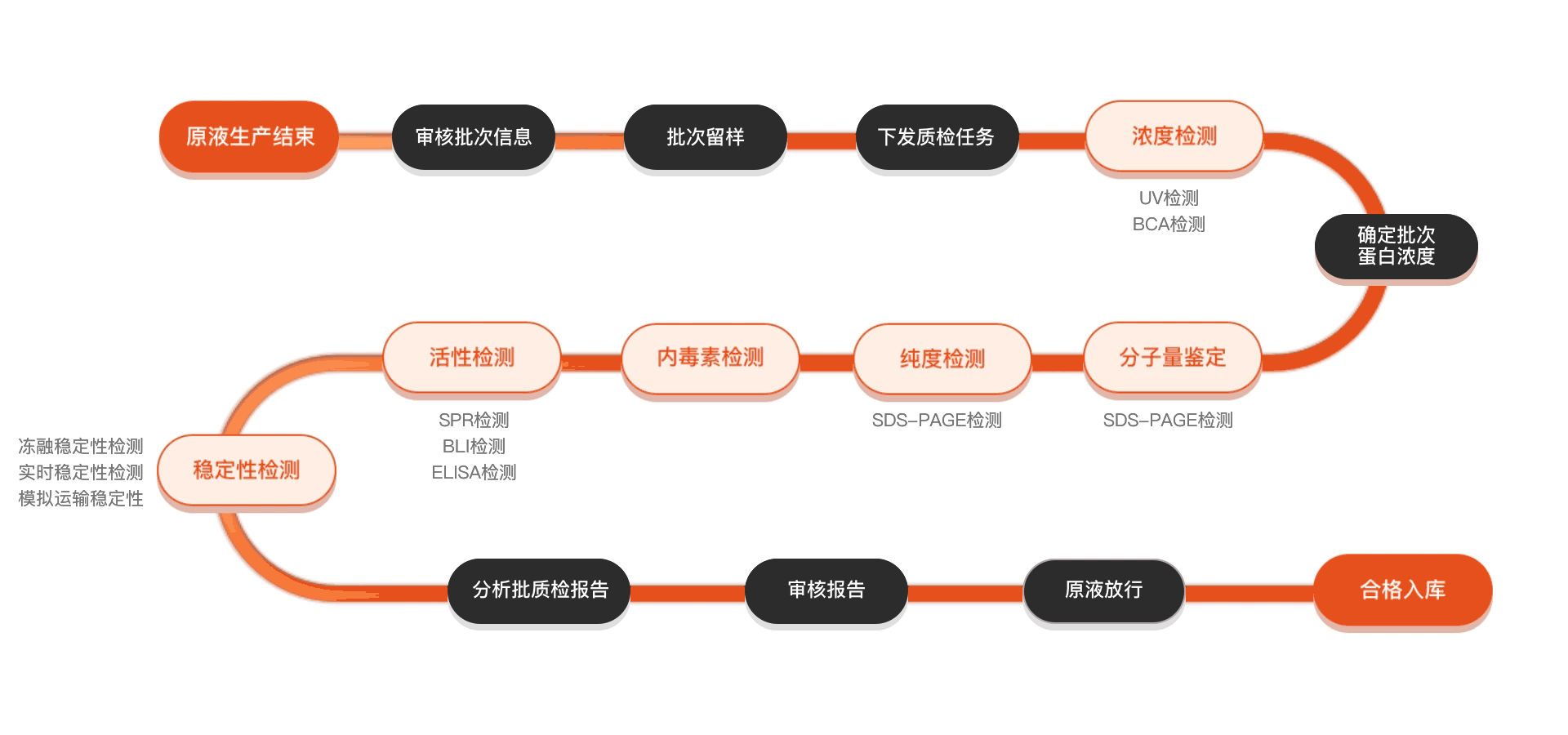
质检流程
相关产品






背景信息
Nectin-1, a member of the immunoglobulin superfamily, is a crucial cell adhesion molecule known for its roles in cell-cell adhesion and epithelium integrity. It functions as a receptor for various pathogens, including herpes simplex virus (HSV), facilitating viral entry into host cells. Given its significance in viral pathogenesis and the immune response, Nectin-1 has become a target of interest in therapeutic research. Researchers aim to develop Nectin-1 recombinant proteins to understand its structure-function relationships, immunogenic properties, and potential as a biomarker for diseases. Furthermore, these recombinant proteins can serve as tools for studying cellular interactions and for developing vaccines against HSV and other pathogens. By elucidating Nectin-1's role in inflammation and immune regulation, scientists hope to harness this knowledge for innovative treatments in virology and immunology. The ongoing exploration of Nectin-1 recombinant proteins may yield promising insights into novel therapeutic strategies, highlighting their potential in addressing infectious diseases and improving human health.