关键信息
-
基因名
IL-17RA
-
简介
IL-17RA (Interleukin 17 receptor A), a receptor for IL-17A and IL-17F, is a type I membrane glycoprotein. It is expressed ubiquitously and exhibits a broad tissue distribution, and plays a role in many inflammatory and autoimmune diseases. IL-17RA is a common co-receptor subunit for other members of the IL-17 family. IL-17RA associates with IL-17RC to form a signaling receptor complex for IL-17A and IL-17F[1][2]. IL-17RA Protein, Human (HEK293, His) is produced in HEK293 cells with six C-Terminal His-tags.
- 应用
-
别名
ADP-ribosyl cyclase 1; cyclic ADP-ribose hydrolase; CD38; T10
-
种属
Human
-
表达系统
HEK293
-
标签
C-6*His
-
纯度
Greater than 90% as determined by SDS-PAGE.
-
蛋白编号
Q96F46-1
-
表达区间
L33-W320
-
氨基酸序列
LRLLDHRALVCSQPGLNCTVKNSTCLDDSWIHPRNLTPSSPKDLQIQLHFAHTQQGDLFPVAHIEWTLQTDASILYLEGAELSVLQLNTNERLCVRFEFLSKLRHHHRRWRFTFSHFVVDPDQEYEVTVHHLPKPIPDGDPNHQSKNFLVPDCEHARMKVTTPCMSSGSLWDPNITVETLEAHQLRVSFTLWNESTHYQILLTSFPHMENHSCFEHMHHIPAPRPEEFHQRSNVTLTLRNLKGCCRHQVQIQPFFSSCLNDCLRHSATVSCPEMPDTPEPIPDYMPLW
-
蛋白长度
Extracellular Domain
-
分子量
52-60 kDa
-
内毒素
< 1.0 EU per μg protein as determined by the LAL method.
-
性状
Freeze-dried powder
-
缓冲液
PBS, pH7.4, containing 0.01% SKL, 1mM DTT, 5% Trehalose and Proclin300.
-
复溶方法
Reconstitute in ddH2O to a concentration of 0.1-0.5 mg/mL. Do not vortex.
- 个性化定制
-
稳定性测试
The thermal stability is described by the loss rate. The loss rate was determined by accelerated thermal degradation test, that is, incubate the protein at 37℃ for 48h, and no obvious degradation and precipitation were observed. The loss rate isless than 8% within the expiration date under appropriate storage condition.
-
保存条件 & 期限
Samples are stable for up to twelve months from date of receipt at -20℃ to -80℃. Store it under sterile conditions at -20℃ to -80℃. It is recommended that the protein be aliquoted for optimal storage. Avoid repeated freeze-thaw cycles.
-
运输条件
In general, recombinant proteins are supplied as lyophilized powder and shipped at ambient temperature. For bulk packages, the proteins are provided as frozen liquid and shipped with blue ice, unless otherwise requested by the customer.
质检流程
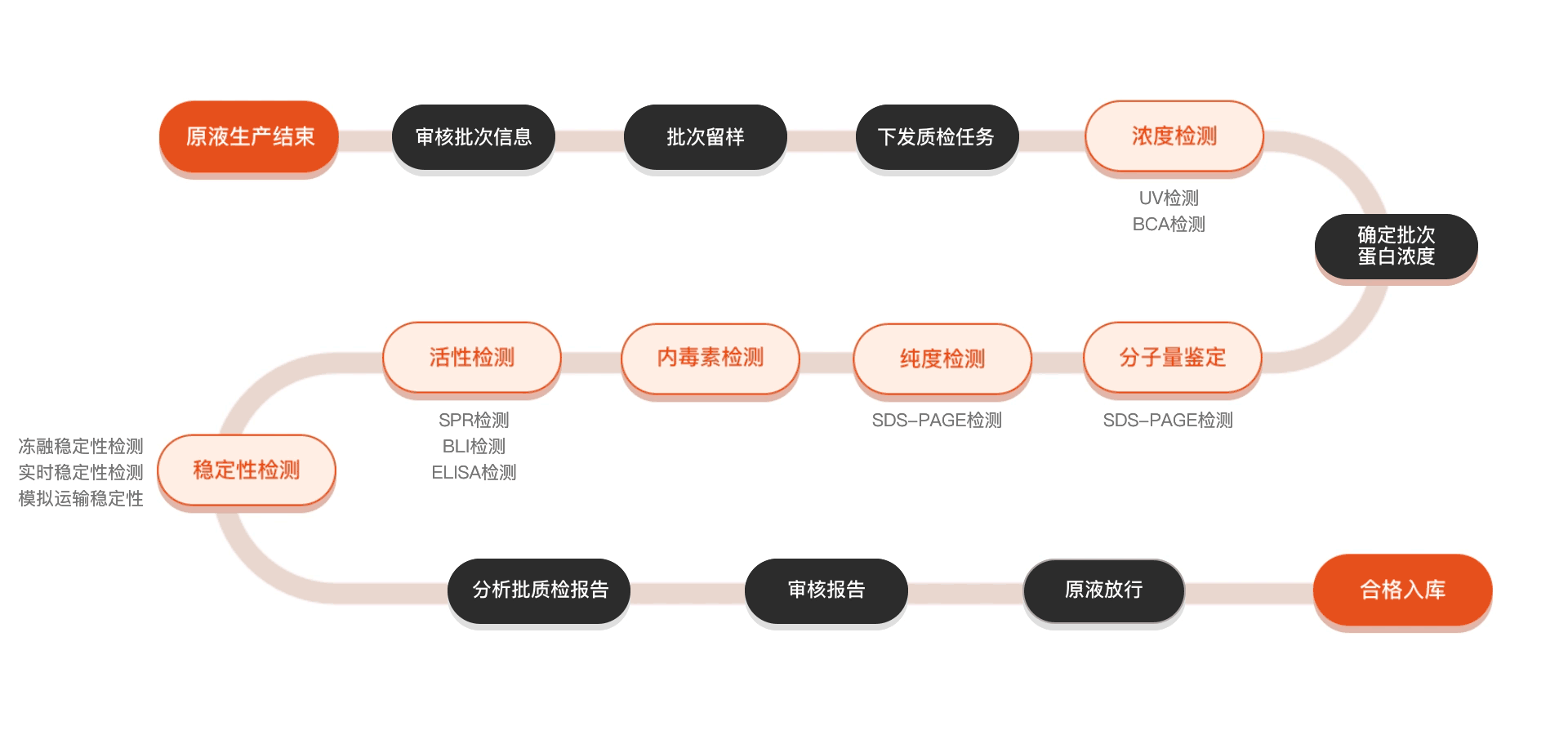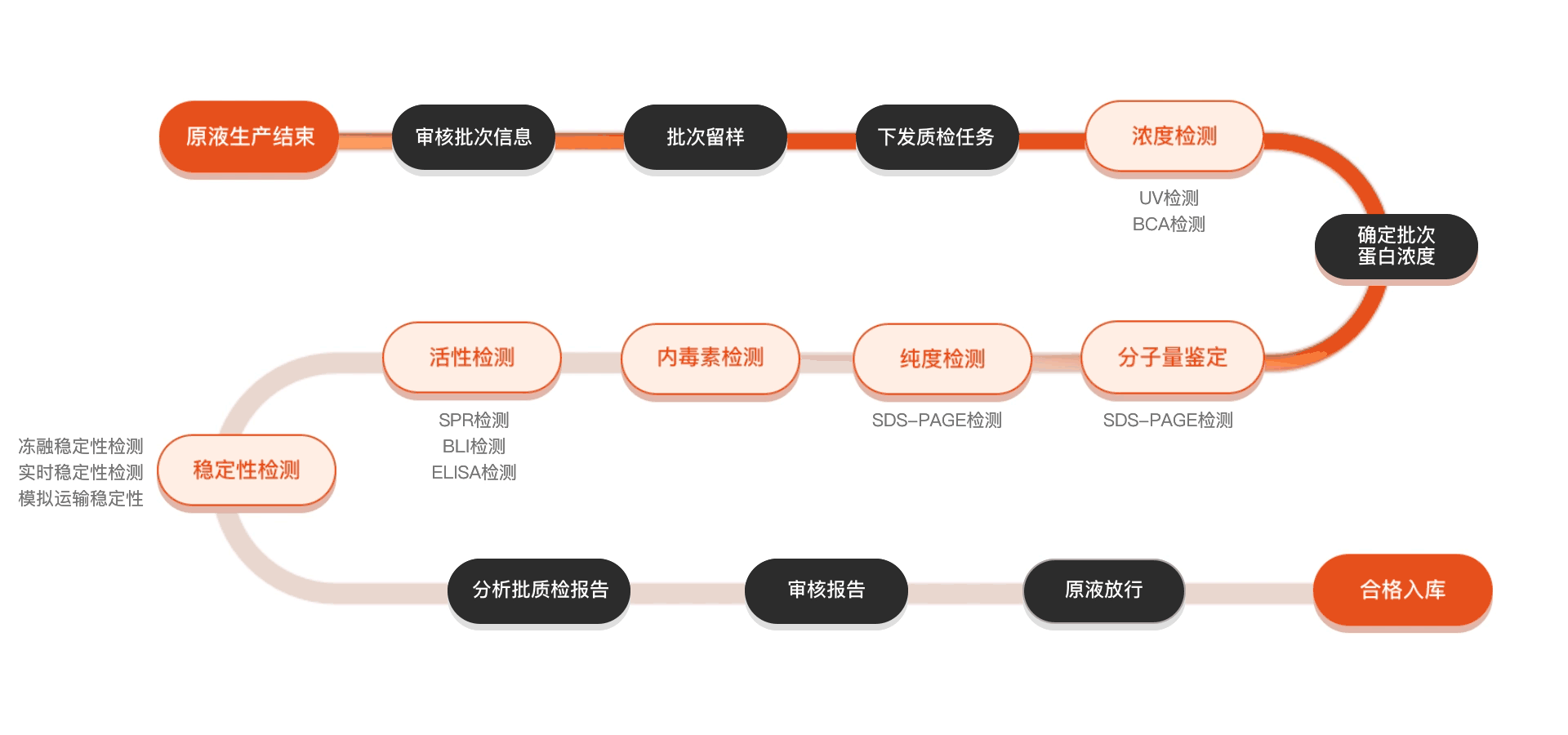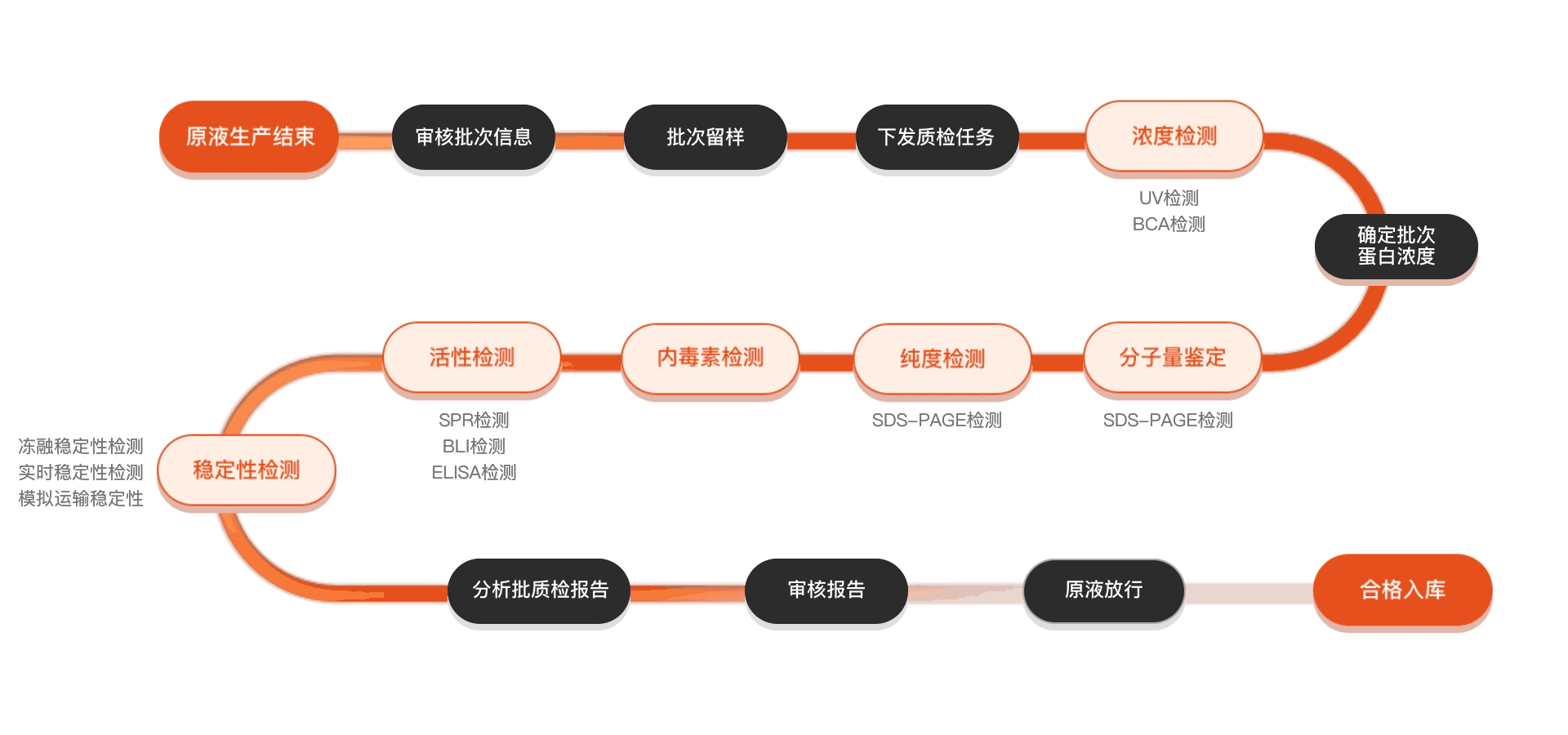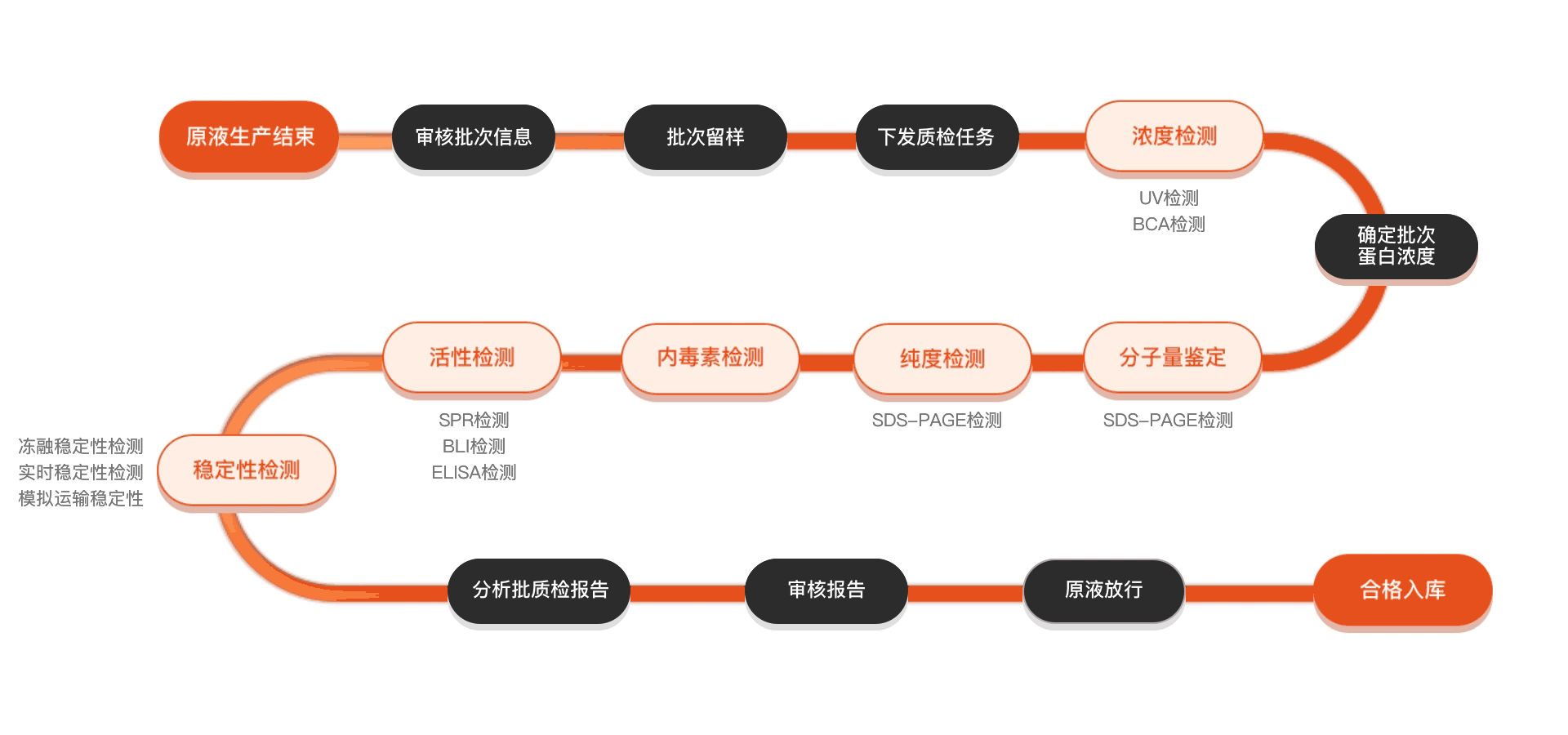
相关产品






背景信息
Interleukin-17 receptor A (IL-17RA) is a crucial component of the IL-17 signaling pathway, which plays a significant role in mediating inflammatory responses and is implicated in various autoimmune diseases, such as rheumatoid arthritis, psoriasis, and multiple sclerosis. The IL-17 family of cytokines, which includes IL-17A, IL-17F, and others, binds to IL-17RA to activate downstream signaling cascades that result in the production of pro-inflammatory cytokines and chemokines. Due to its pivotal role in the immune response, IL-17RA has emerged as a promising therapeutic target. Research into recombinant protein forms of IL-17RA aims to better understand its structure, function, and interaction with other molecules within the immune system. Recombinant IL-17RA proteins can be utilized to develop biologics that inhibit these pathways, potentially providing new treatment options for patients suffering from IL-17-mediated diseases. Additionally, studying IL-17RA may reveal insights into its involvement in normal immune function and help delineate its contributions to disease pathogenesis. Thus, the exploration of IL-17RA recombinant proteins not only enhances our understanding of immune regulation but also holds significant promise for innovative therapeutic strategies in managing a range of inflammatory disorders.