关键信息
-
基因名
PD-L1
-
简介
The PD-L1 protein critically regulates immune tolerance by acting as a ligand for PDCD1/PD-1, regulating T cell activation threshold, and limiting effector responses. It may act as a costimulatory molecule for IL10-producing T cell subsets. PD-L1 Protein, Human (HEK293, Flag) is the recombinant human-derived PD-L1 protein, expressed by HEK293 , with C-Flag labeled tag.
- 应用
-
别名
Programmed Cell Death 1 Ligand 1; PD-L1; PDCD1 Ligand 1; Programmed Death Ligand 1; B7 Homolog 1; B7-H1; CD274; B7H1; PDCD1L1; PDCD1LG1; PDL1
-
种属
Human
-
表达系统
HEK293
-
标签
C-Flag
-
纯度
Greater than 90% as determined by SDS-PAGE.
-
蛋白编号
Q9NZQ7-1
-
表达区间
F19-T239
-
氨基酸序列
FTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYNKINQRILVVDPVTSEHELTCQAEGYPKAEVIWTSSDHQVLSGKTTTTNSKREEKLFNVTSTLRINTTTNEIFYCTFRRLDPEENHTAELVIPELPLAHPPNERT
-
蛋白长度
Partial
-
分子量
35-40 kDa
-
内毒素
< 1.0 EU per μg protein as determined by the LAL method.
-
性状
Freeze-dried powder
-
缓冲液
PBS, pH7.4, containing 0.01% SKL, 1mM DTT, 5% Trehalose and Proclin300.
-
复溶方法
Reconstitute in ddH2O to a concentration of 0.1-0.5 mg/mL. Do not vortex.
- 个性化定制
-
稳定性测试
The thermal stability is described by the loss rate. The loss rate was determined by accelerated thermal degradation test, that is, incubate the protein at 37℃ for 48h, and no obvious degradation and precipitation were observed. The loss rate isless than 8% within the expiration date under appropriate storage condition.
-
保存条件 & 期限
Samples are stable for up to twelve months from date of receipt at -20℃ to -80℃. Store it under sterile conditions at -20℃ to -80℃. It is recommended that the protein be aliquoted for optimal storage. Avoid repeated freeze-thaw cycles.
-
运输条件
In general, recombinant proteins are supplied as lyophilized powder and shipped at ambient temperature. For bulk packages, the proteins are provided as frozen liquid and shipped with blue ice, unless otherwise requested by the customer.
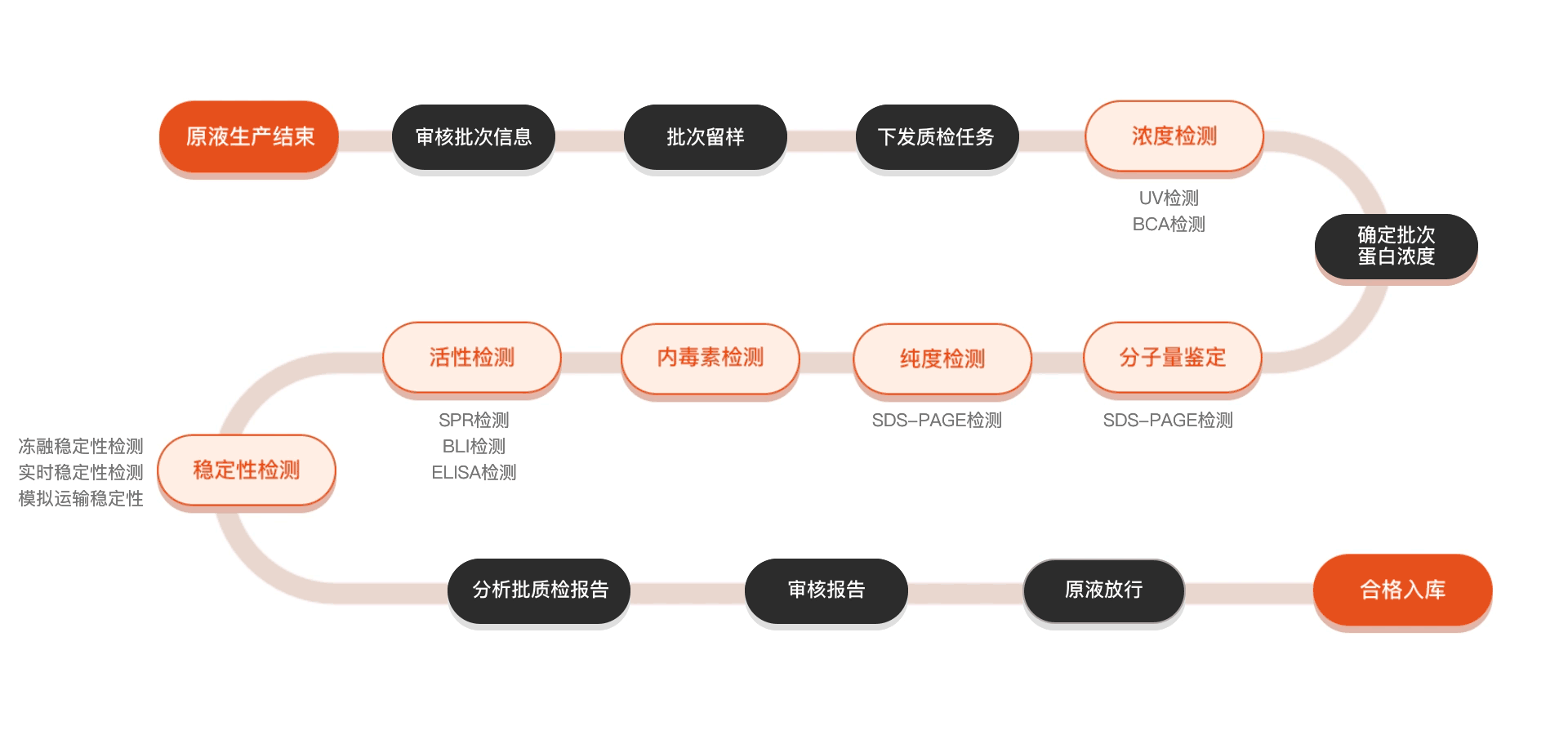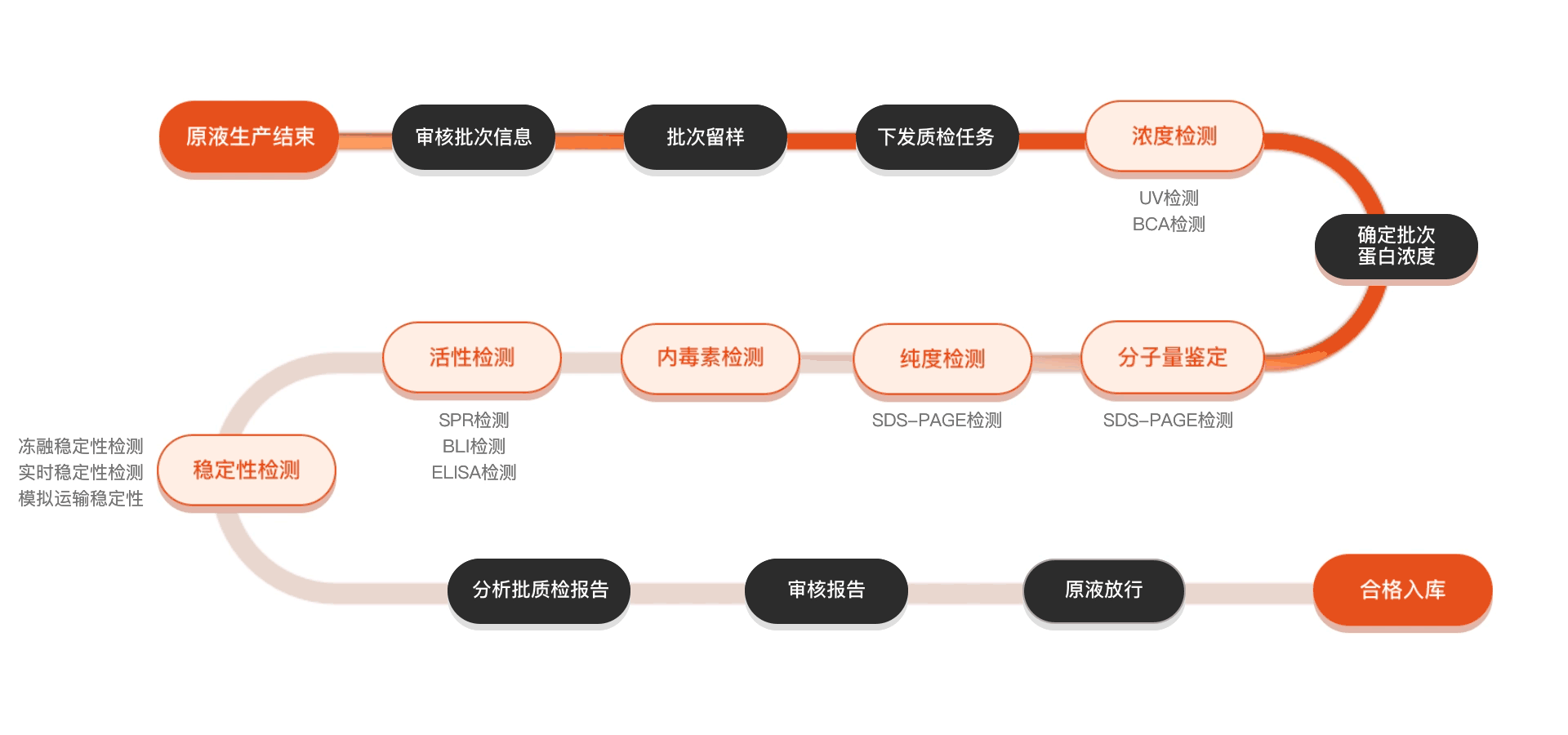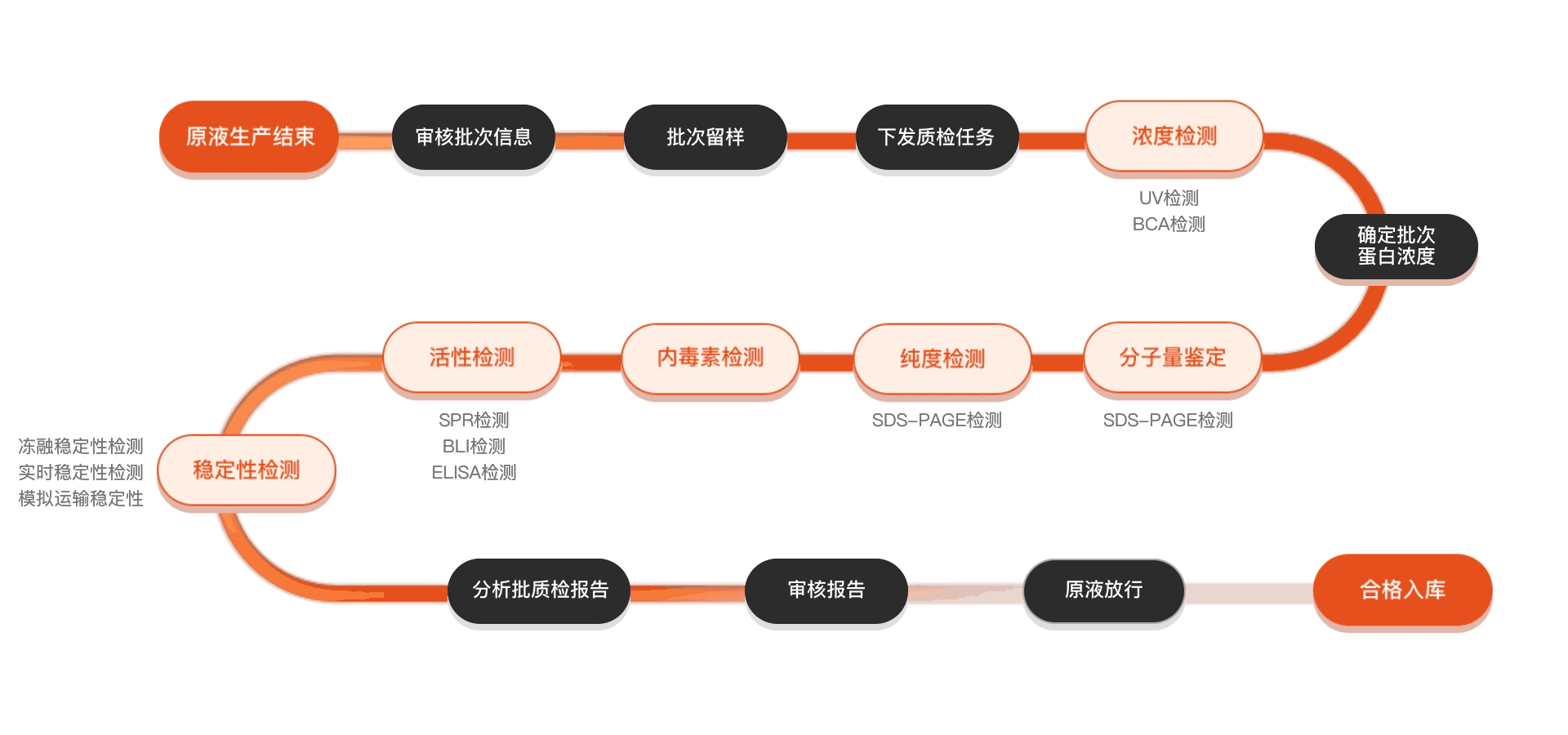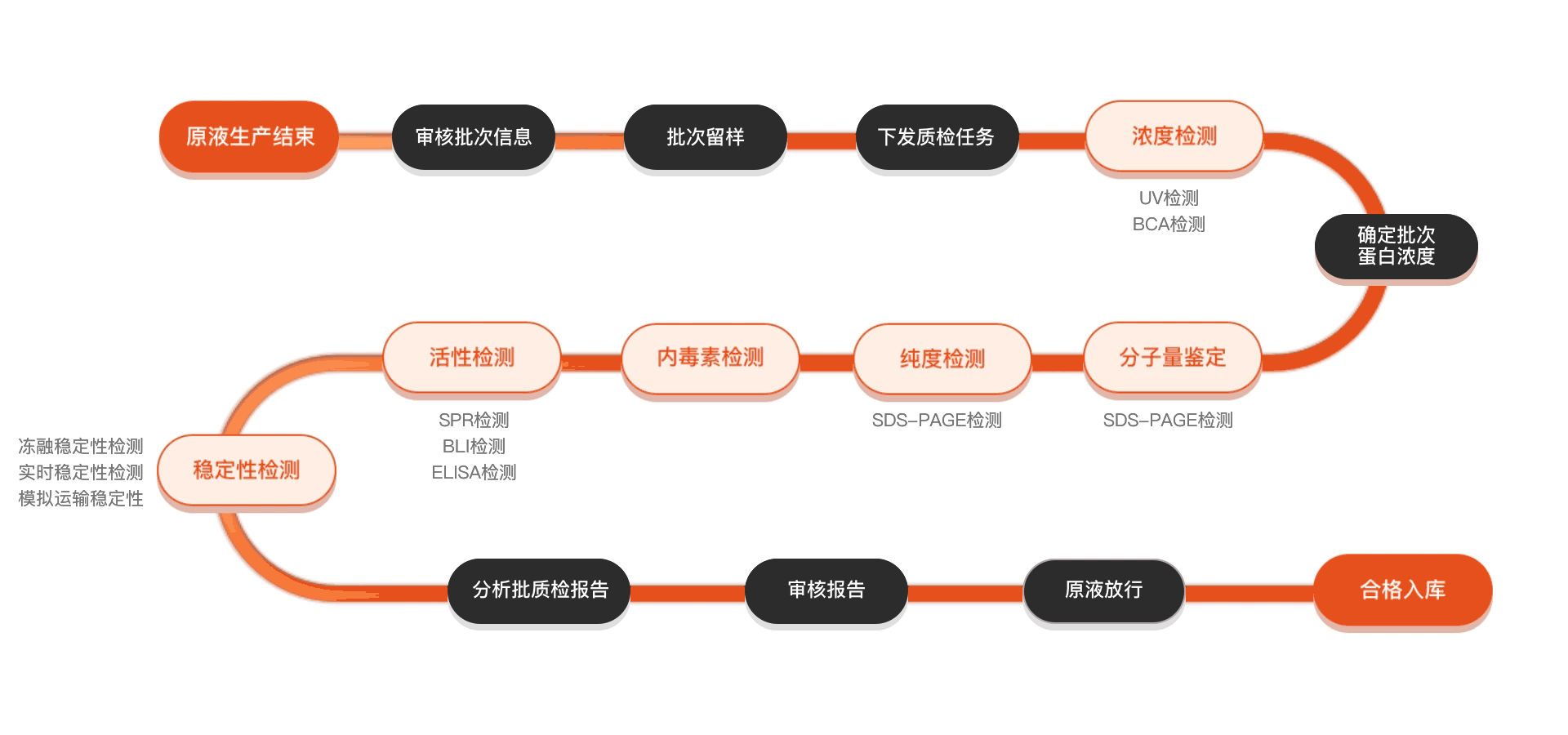
质检流程
相关产品






背景信息
PD-L1 (Programmed Death-Ligand 1) is a critical immune checkpoint molecule that plays a significant role in regulating the immune response, particularly in the context of cancer. It is primarily expressed on the surface of tumor cells and interacts with the PD-1 receptor on T cells, leading to the inhibition of T cell activation and proliferation. This mechanism is often exploited by tumors to evade immune detection and destruction. Due to its pivotal role in immune regulation, PD-L1 has become a prominent target for cancer immunotherapy, with the development of monoclonal antibodies that block this interaction, thereby enhancing anti-tumor immunity. Research on recombinant PD-L1 proteins has gained momentum as scientists seek to better understand its structure, function, and the dynamics of its interactions with other immune molecules. These studies provide insights into the potential therapeutic applications of PD-L1 inhibitors and the design of more effective immunotherapeutic strategies. Moreover, the production of recombinant PD-L1 proteins allows for detailed biochemical and biophysical characterizations, facilitating the identification of specific binding affinities and the delineation of signaling pathways involved in immune regulation. Overall, the continual exploration of PD-L1 and its role in the immune system underscores its importance in developing new cancer therapies aimed at reactivating the body’s own immune response against malignancies.