关键信息
-
基因名
POF1B
- 应用
-
别名
2310066B14Rik; FLJ22792; POF; Pof1b; POF1B_HUMAN; POF2B; Premature ovarian failure protein 1B; Protein POF1B; RGD1560798; RP1-75N13.2; RP23-336F13.1
-
种属
Human
-
表达系统
E. coli
-
标签
His tag N-Terminus
-
纯度
Greater than 90% as determined by SDS-PAGE.
-
蛋白编号
Q8WVV4
-
表达区间
1-589 aa
-
氨基酸序列
MSSSYWSETS SSSCGTQQLP EVLQCQPQHY HCYHQSSQAQ QPPEKNVVYE RVRTYSGPMN KVVQALDPFN SREVLSPLKT TSSYQNLVWS DHSQELHSPT LKISTCAPST LHITQNTEQE LHSPTVKLTT YPQTTIRKYV VQNPEQEPLS QFLRGSHFFP GNNVIYEKTI RKVEKLNTDQ GCHPQAQCHH HIIQQPQVIH SAHWQQPDSS QQIQAITGNN PISTHIGNEL CHSGSSQICE QVIIQDDGPE KLDPRYFGEL LADLSRKNTD LYHCLLEHLQ RIGGSKQDFE STDESEDIES LIPKGLSEFT KQQIRYILQM RGMSDKSLRL VLSTFSNIRE ELGHLQNDMT SLENDKMRLE KDLSFKDTQL KEYEELLASV RANNHQQQQG LQDSSSKCQA LEENNLSLRH TLSDMEYRLK ELEYCKRNLE QENQNLRMQV SETCTGPMLQ AKMDEIGNHY TEMVKNLRME KDREICRLRS QLNQYHKDVS KREGSCSDFQ FKLHELTSLL EEKDSLIKRQ SEELSKLRQE IYSSHNQPST GGRTTITTKK YRTQYPILGL LYDDYEYIPP GSETQTIVIE KTEDKYTCP
-
分子量
68.0 kDa
-
内毒素
< 1.0 EU per μg protein as determined by the LAL method.
-
性状
Freeze-dried powder
-
缓冲液
PBS, pH7.4, containing 0.01% SKL, 1mM DTT, 5% Trehalose and Proclin300.
-
复溶方法
Reconstitute in ddH2O to a concentration of 0.1-0.5 mg/mL. Do not vortex.
- 个性化定制
-
稳定性测试
The thermal stability is described by the loss rate. The loss rate was determined by accelerated thermal degradation test, that is, incubate the protein at 37℃ for 48h, and no obvious degradation and precipitation were observed. The loss rate isless than 8% within the expiration date under appropriate storage condition.
-
保存条件 & 期限
Samples are stable for up to twelve months from date of receipt at -20℃ to -80℃. Store it under sterile conditions at -20℃ to -80℃. It is recommended that the protein be aliquoted for optimal storage. Avoid repeated freeze-thaw cycles.
-
运输条件
In general, recombinant proteins are supplied as lyophilized powder and shipped at ambient temperature. For bulk packages, the proteins are provided as frozen liquid and shipped with blue ice, unless otherwise requested by the customer.
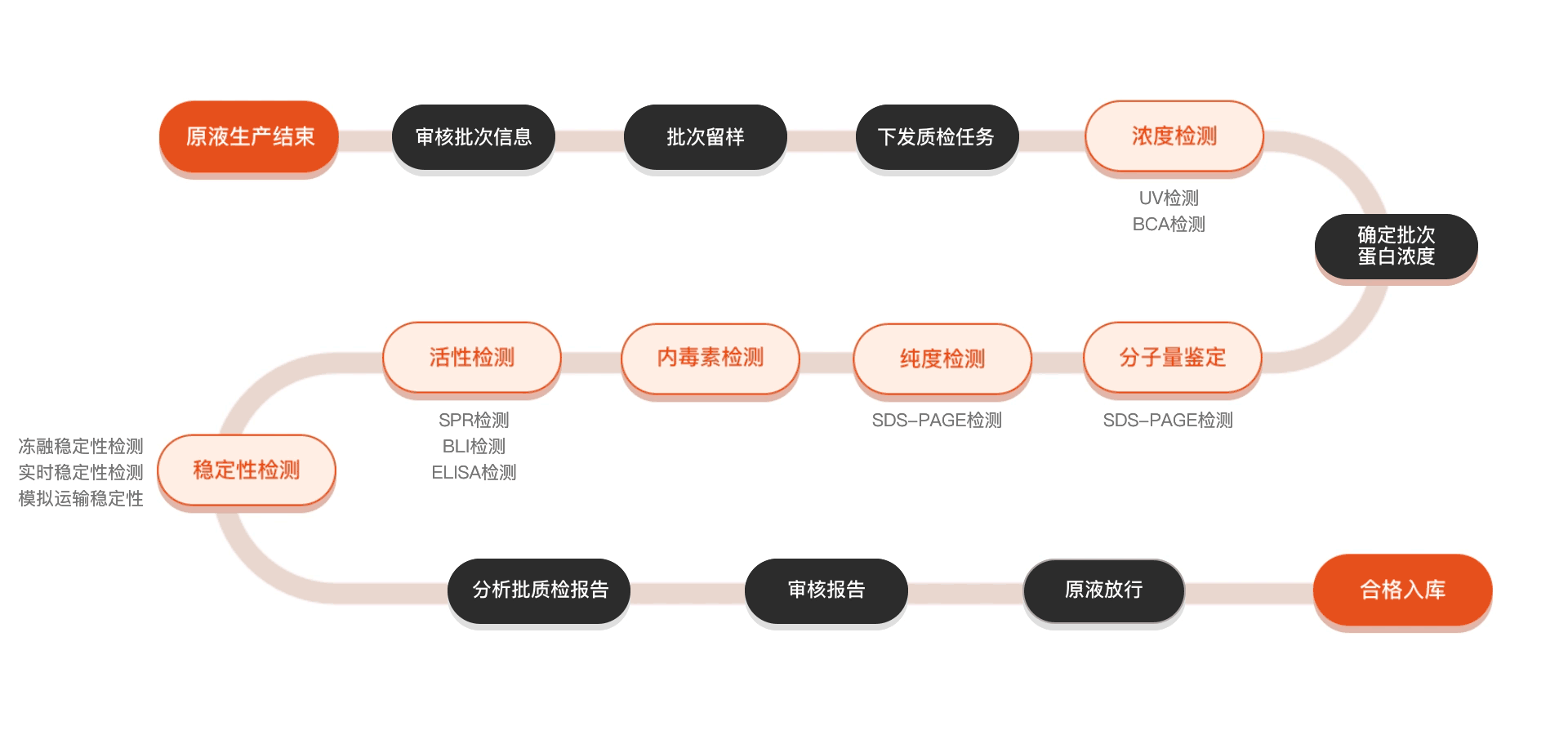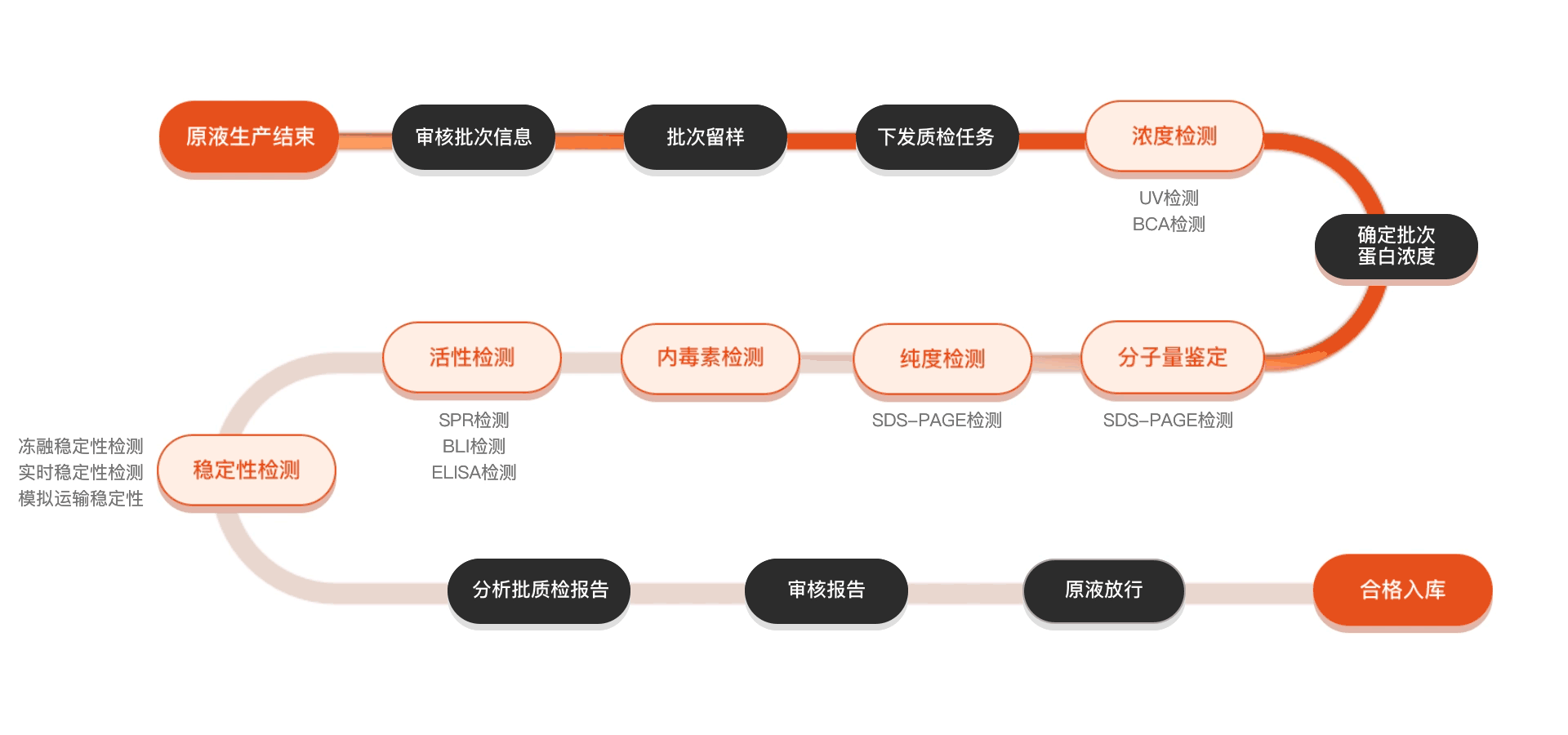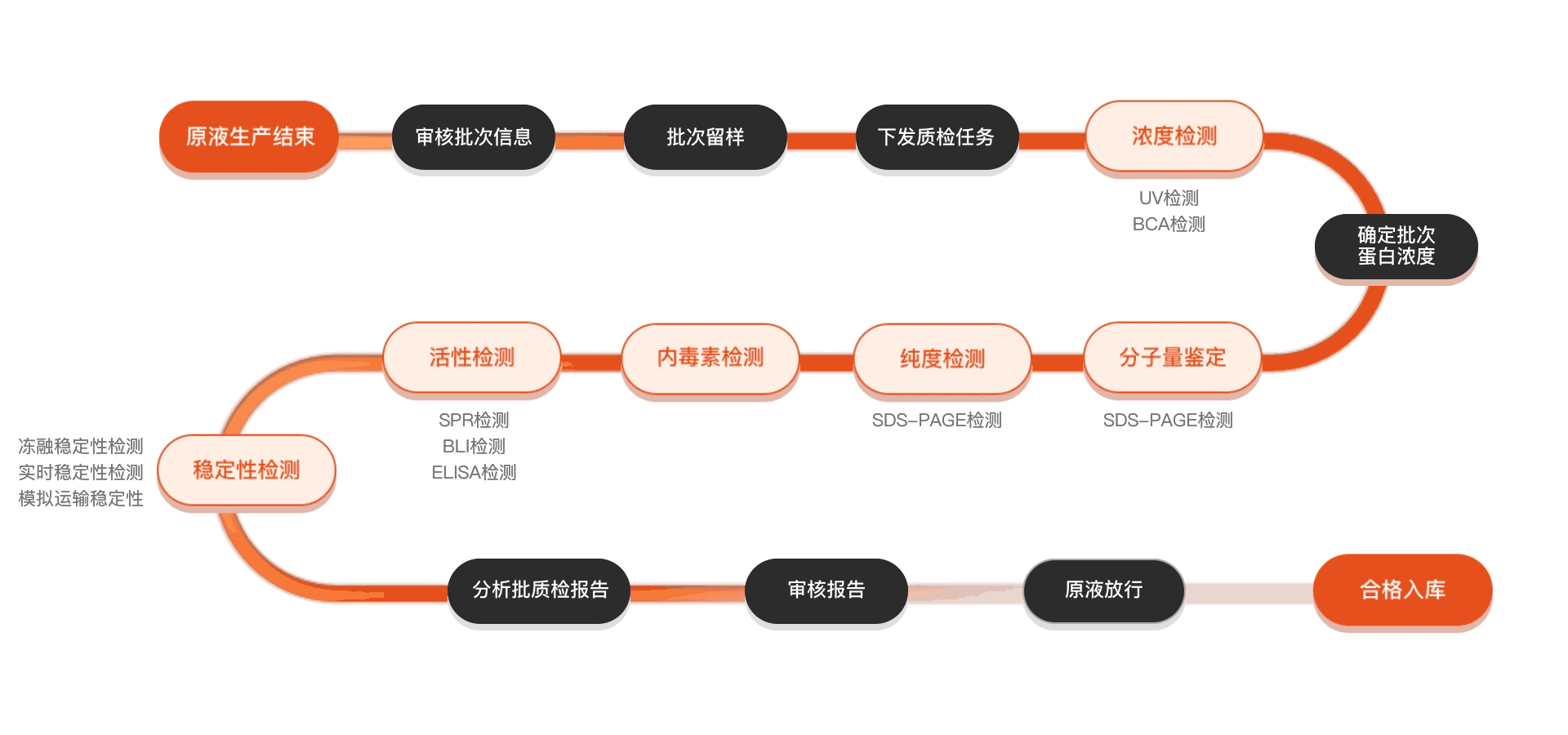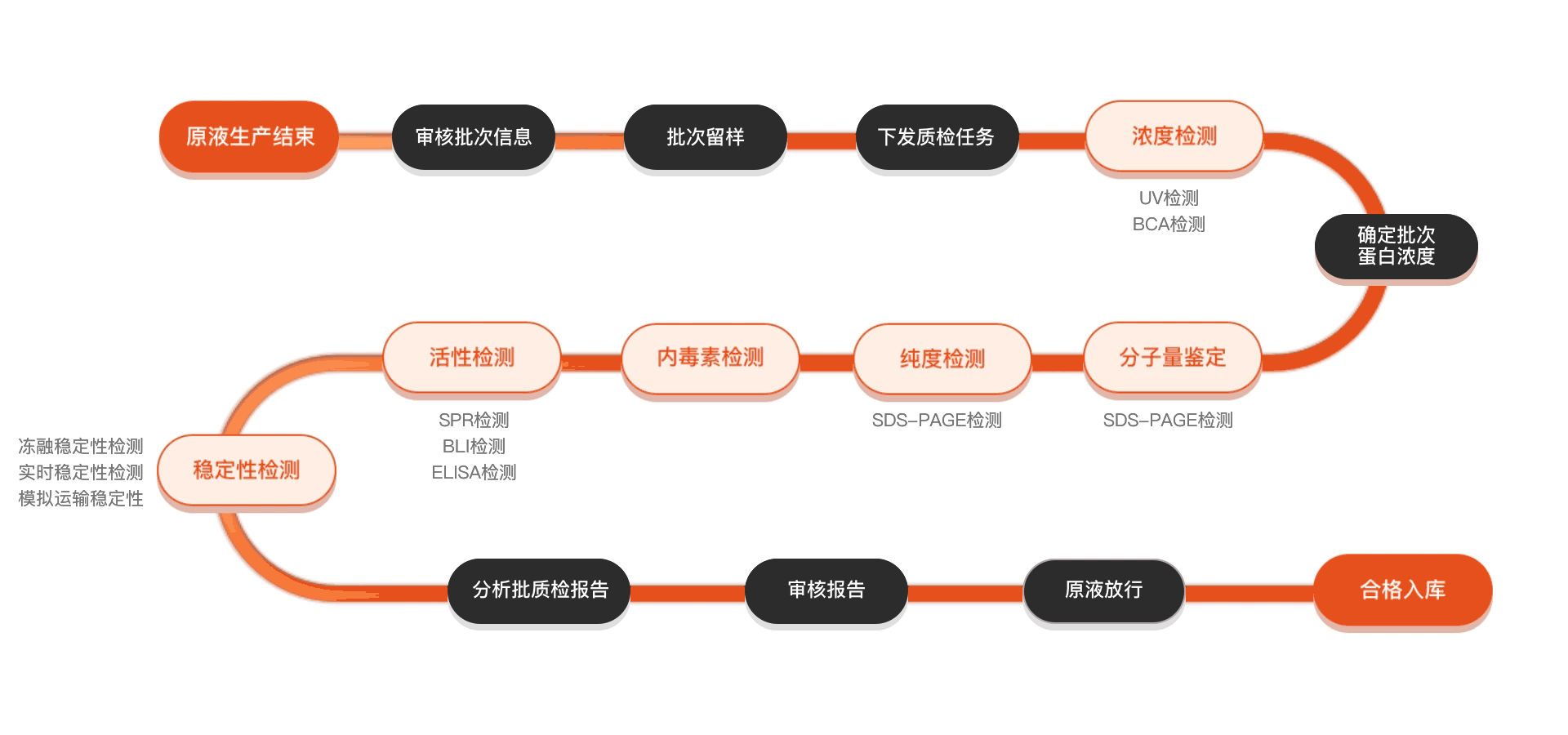
质检流程
相关产品






背景信息
POF1B, or POF1B protein, has garnered significant attention in the field of molecular biology and genetics due to its critical role in cellular processes. Initially identified as a protein involved in cell signaling and regulation, POF1B is particularly relevant in the context of developmental biology and disease. Research has revealed that mutations in the POF1B gene are associated with specific developmental disorders, particularly those affecting the craniofacial region, highlighting its importance in human health. The study of POF1B at the molecular level is crucial for understanding its functional mechanisms and interactions within cellular pathways. Furthermore, the investigation of POF1B as a recombinant protein has great implications for therapeutic applications, especially in gene therapy and regenerative medicine. Through advanced techniques in protein engineering and expression systems, researchers aim to produce POF1B in a laboratory setting, allowing for a deeper exploration of its structure and function. Understanding the role of POF1B not only contributes to our knowledge of genetic disorders but also paves the way for potential interventions and treatments that could ameliorate the effects of mutations associated with this protein. As research progresses, POF1B continues to be a focal point for scientists aiming to unravel the complexities of genetic regulation and its impact on human development.