关键信息
-
基因名
DPP4
- 应用
-
别名
DPP4;ADCP2;CD26;Dipeptidyl peptidase 4
-
种属
Human
-
表达系统
HEK293 Cell
-
标签
N-terminal Fc-Tag
-
纯度
Greater than 90% as determined by SDS-PAGE.
-
蛋白编号
P27487
-
表达区间
29-766aa
-
氨基酸序列
NKGTDDATADSRKTYTLTDYLKNTYRLKLYSLRWISDHEYLYKQENNILVFNAE YGNSSVFLENSTFDEFGHSINDYSISPDGQFILLEYNYVKQWRHSYTASYDIYD LNKRQLITEERIPNNTQWVTWSPVGHKLAYVWNNDIYVKIEPNLPSYRITWTGK EDIIYNGITDWVYEEEVFSAYSALWWSPNGTFLAYAQFNDTEVPLIEYSFYSDE SLQYPKTVRVPYPKAGAVNPTVKFFVVNTDSLSSVTNATSIQITAPASMLIGDH YLCDVTWATQERISLQWLRRIQNYSVMDICDYDESSGRWNCLVARQHIEMSTTG WVGRFRPSEPHFTLDGNSFYKIISNEEGYRHICYFQIDKKDCTFITKGTWEVIG IEALTSDYLYYISNEYKGMPGGRNLYKIQLSDYTKVTCLSCELNPERCQYYSVS FSKEAKYYQLRCSGPGLPLYTLHSSVNDKGLRVLEDNSALDKMLQNVQMPSKKL DFIILNETKFWYQMILPPHFDKSKKYPLLLDVYAGPCSQKADTVFRLNWATYLA STENIIVASFDGRGSGYQGDKIMHAINRRLGTFEVEDQIEAARQFSKMGFVDNK RIAIWGWSYGGYVTSMVLGSGSGVFKCGIAVAPVSRWEYYDSVYTERYMGLPTP EDNLDHYRNSTVMSRAENFKQVEYLLIHGTADDNVHFQQSAQISKALVDVGVDF QAMWYTDEDHGIASSTAHQHIYTHMSHFIKQCFSLP
-
分子量
112.0kDa
-
内毒素
< 1.0 EU per μg protein as determined by the LAL method.
-
性状
Freeze-dried powder
-
缓冲液
PBS, pH7.4, 5% Trehalose
-
复溶方法
Reconstitute in ddH2O to a concentration of 0.1-0.5 mg/mL. Do not vortex.
- 个性化定制
-
稳定性测试
The thermal stability is described by the loss rate. The loss rate was determined by accelerated thermal degradation test, that is, incubate the protein at 37℃ for 48h, and no obvious degradation and precipitation were observed. The loss rate isless than 8% within the expiration date under appropriate storage condition.
-
保存条件 & 期限
Samples are stable for up to twelve months from date of receipt at -20℃ to -80℃. Store it under sterile conditions at -20℃ to -80℃. It is recommended that the protein be aliquoted for optimal storage. Avoid repeated freeze-thaw cycles.
-
运输条件
In general, recombinant proteins are supplied as lyophilized powder and shipped at ambient temperature. For bulk packages, the proteins are provided as frozen liquid and shipped with blue ice, unless otherwise requested by the customer.
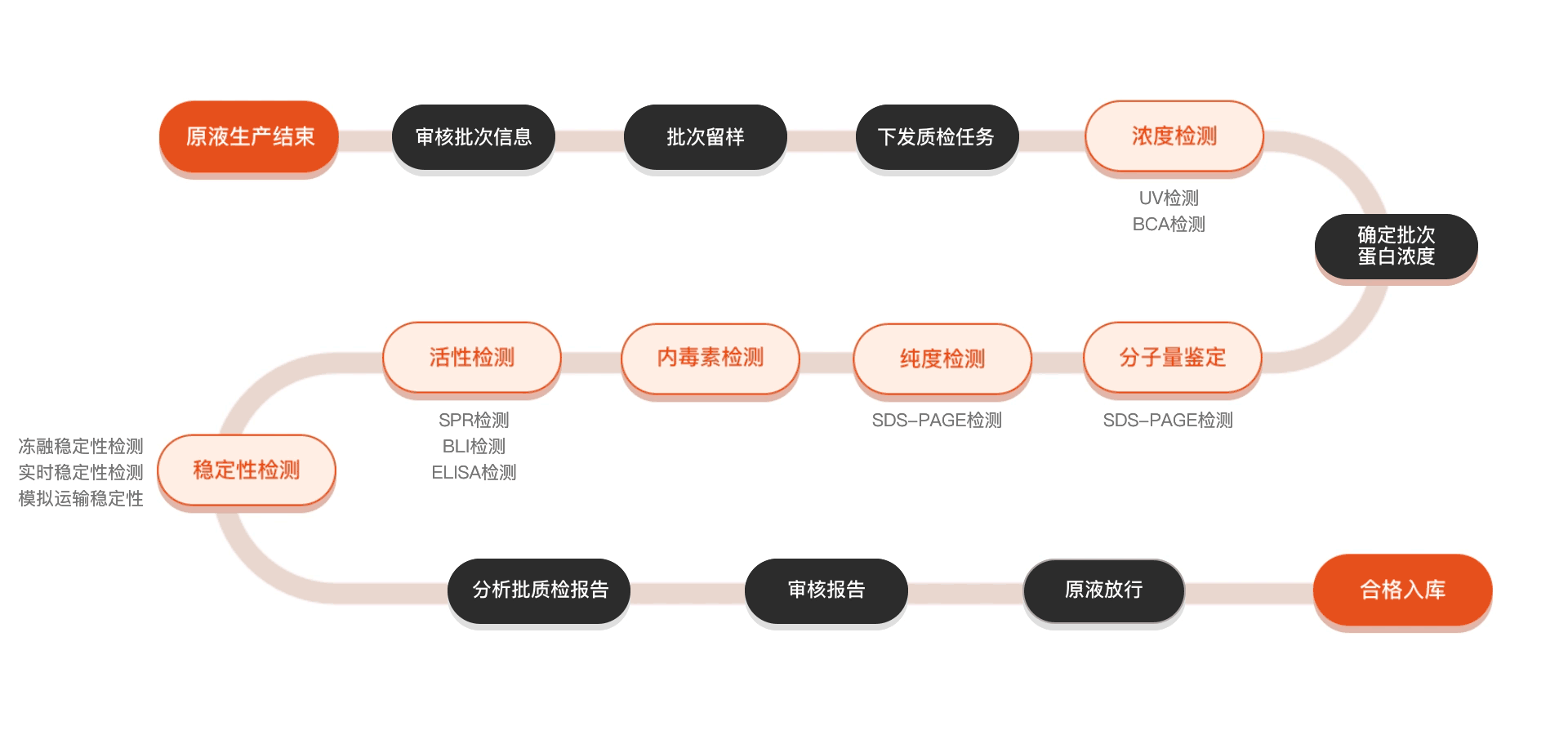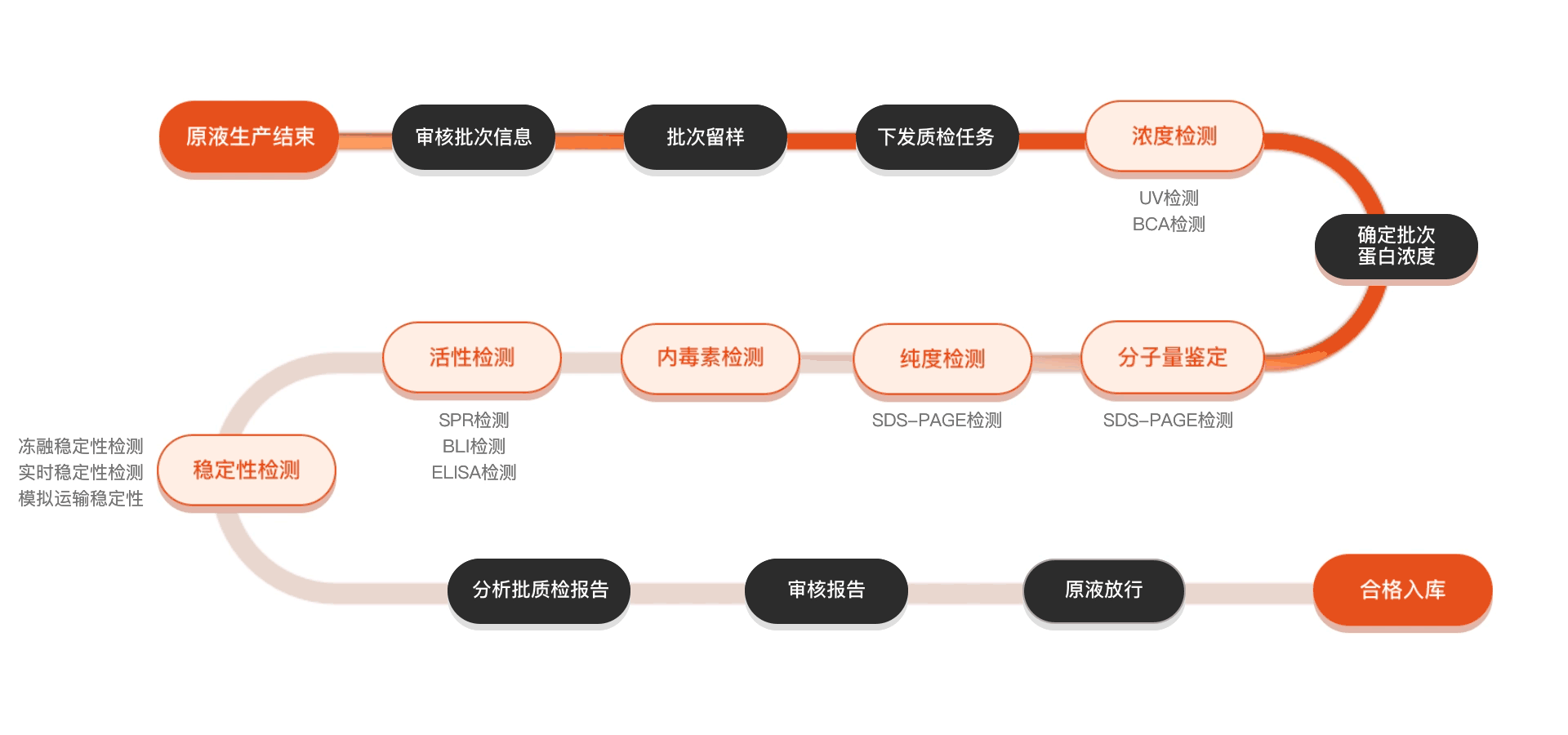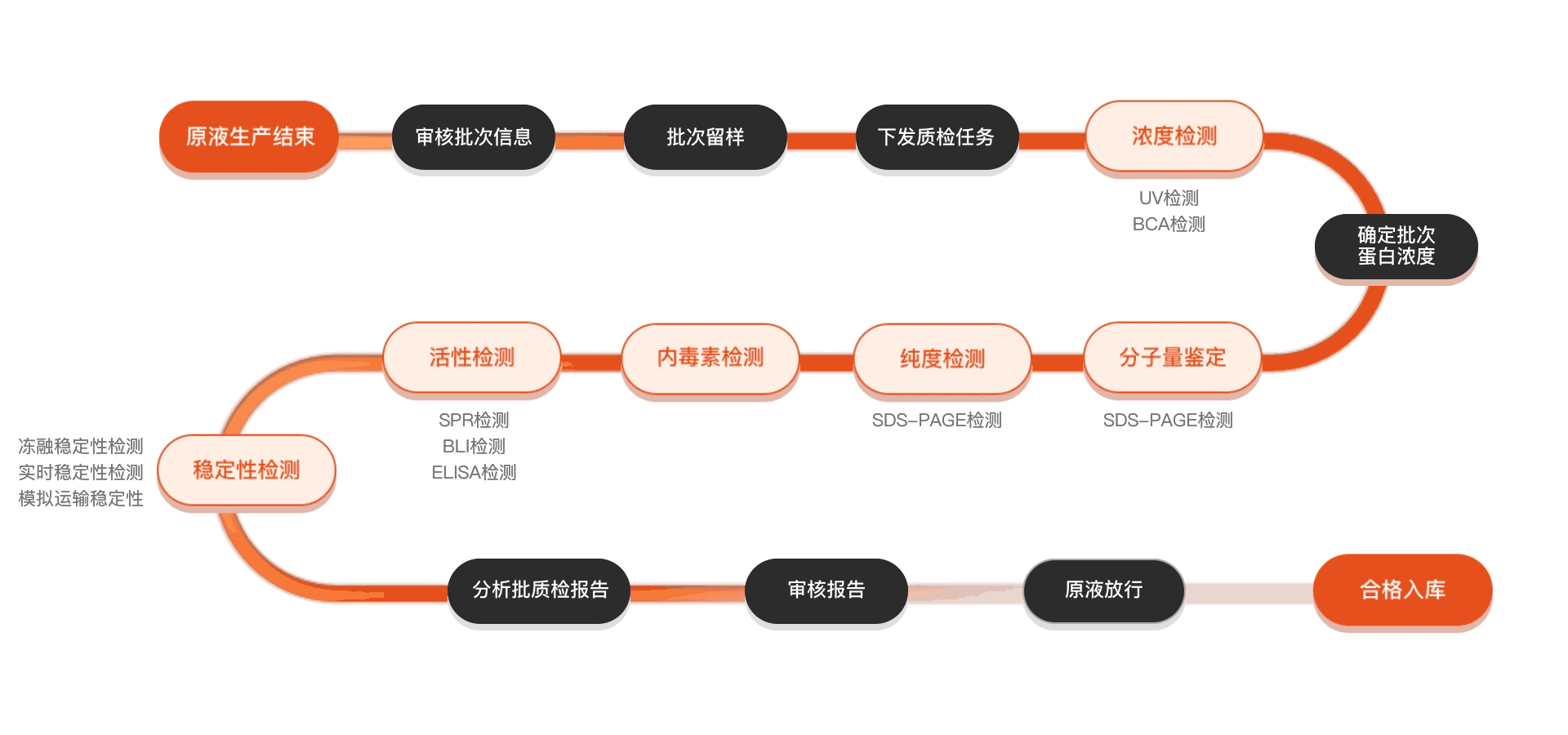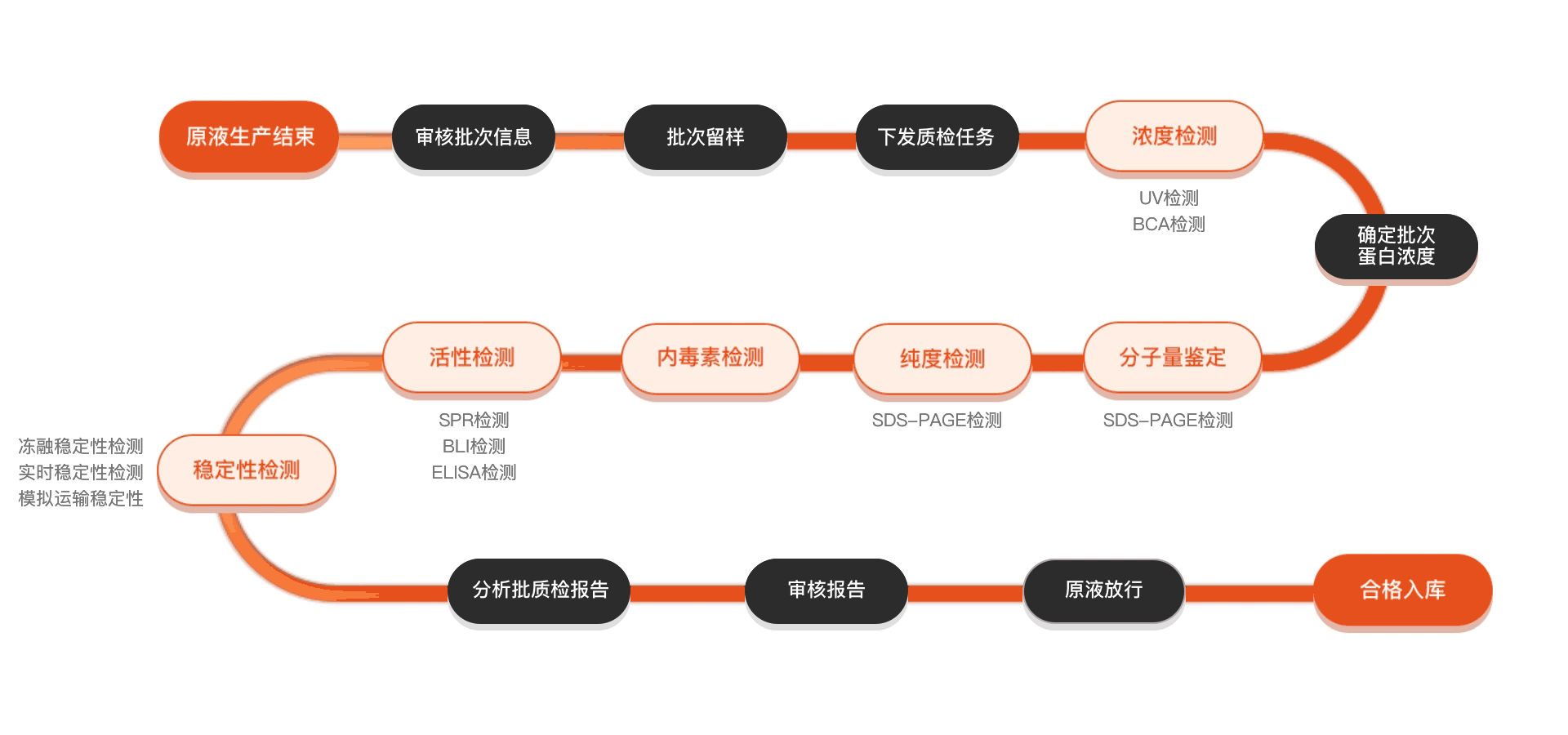
质检流程
相关产品






背景信息
Dipeptidyl peptidase-4 (DPP4) is a serine protease that plays a crucial role in glucose metabolism and immune regulation. Initially identified as an enzyme that inactivates incretin hormones, such as GLP-1 and GIP, which are vital for insulin secretion and glucose homeostasis, DPP4 has gained attention in various fields of research. Elevated DPP4 activity has been implicated in metabolic disorders, including type 2 diabetes and obesity, making it a potential therapeutic target. Additionally, DPP4 is expressed on various immune cells, suggesting its involvement in immune responses and inflammation. Recent studies have indicated that DPP4 might also play a role in cancer biology, as its activity can influence tumor cell proliferation and migration. The development of DPP4 inhibitors has proven beneficial in managing type 2 diabetes, leading to the exploration of DPP4 as a biomarker for disease progression and prognosis. Furthermore, investigations into DPP4 as a recombinant protein have potential applications in understanding its enzymatic mechanisms, its interactions with other proteins, and the development of novel therapeutic strategies. Overall, research on DPP4 and its recombinant forms presents exciting opportunities to deepen our understanding of its multifaceted roles in human health and disease.